Capillary malformation-arteriovenous malformation syndrome is a rare genodermatosis with cutaneous capillary malformations and a risk of associated fast-flow malformations. We describe here a four-generation family with a novel heterozygous pathogenic variant in the EPHB4 gene (NM_004444.5 (EPHB4): c.2224G>C, p.(Ala742Pro)). A review of the literature retrieved 127 patients with capillary malformation-arteriovenous malformation syndrome and confirmed variants in EPHB4. Multiple capillary malformations were present in 114 (89.76%) patients, and 12 (9.44%) patients had a solitary capillary malformation. Arteriovenous malformations/fistulas were present in 23 (18.1%) patients, and were located within the central nervous system in 5 (3.9%) patients. Not all papers included description of epistaxis. Telangiectasias were reported in 28 (22%) patients, and Bier spots were described in 20 (15.7%) patients. The clinical characteristics of capillary malformation-arteriovenous malformation syndrome are diverse and often discrete, which can make it difficult to distinguish capillary malformation-arteriovenous malformation syndrome from hereditary haemorrhagic telangiectasia.
Key words: capillary malformation-arteriovenous malformation syndrome; CM-AVM2; EPHB4; arteriovenous malformation.
Accepted Jan 28, 2022; Epub ahead of print Jan 28, 2022
Acta Derm Venereol 2022; 102: adv00662.
DOI: 10.2340/actadv.v102.1126
Corr: Anna Trier Heiberg Brix, Department of Clinical Genetics, Odense University Hospital, J. B. Winsløwsvej 19, 3, DK-5000 Odense, Denmark. E-mail: anna.trier.heiberg.brix@rsyd.dk
SIGNIFICANCE
Capillary malformation-arteriovenous malformation syndrome is a rare inherited disease. A four-generation family with capillary malformation-arteriovenous malformation syndrome is presented, and data from other published capillary malformation-arteriovenous malformation syndrome cases are compiled to establish a more thorough description and estimate of the frequency of capillary malformations, arteriovenous malformations or fistulas, epistaxis, telangiectasia, and Bier spots in this condition. The four-generation family has a novel variant in the EPHB4 gene.
INTRODUCTION
Capillary malformation-arteriovenous malformation (CM-AVM) syndrome is a rare, autosomal dominant disease characterized by multiple cutaneous capillary malformations (CMs) and risk of having 1 or more arteriovenous malformations (AVMs) and/or arteriovenous fistulas (AVFs) (1–5). Eerola et al. named the syndrome CM-AVM in 2003 and, since 2017, CM-AVM has been subdivided into CM-AVM1 (OMIM 608354) and CM-AVM2 (OMIM 618196) based on pathogenic variants in 2 different genes: RASA1 on chromosome 5 and EPHB4 on chromosome 7, respectively (1, 2). An inherited or de novo (25%) heterozygous loss-of-function germline pathogenic variant constitutes the first hit, followed by a second hit inducing a pathogenic variant in the other allele of the endothelial cells (6). This two-hit mechanism is known from other inherited vascular malformations with multifocal appearance (7).
CM-AVM has an almost 100% penetrance, however the clinical characteristics may vary.
Capillary malformation-arteriovenous malformation syndrome-1 and -2 phenotypes
CM-AVM1 and CM-AVM2 have some similar clinical characteristics (Table I). Both conditions present with cutaneous CMs that can be associated with the presence of 1 or more AVMs/AVFs. The CMs are described as macular, possible multifocal, light red, red or violet lesions varying in size typically from 1 to 3 cm in diameter (1–3, 8–11). Larger lesions can occur, and CMs up to 15 cm in diameter have been described (8). A white halo can be seen around the CMs as a result of micro shunting. The cutaneous CMs can be present at birth and new lesions can evolve over time (12, 13). The typical locations are face, thorax and extremities. Doppler pulsation can be found even in minor CMs and is explained by an arterial component in the CMs (14, 15). As a consequence a higher skin temperature or thrill in the affected area can be observed (10). Fast-flow malformations, such as AVMs/AVFs, can be present in different sizes and locations. Cutaneous, subcutaneous, intramuscular, intraosseous, intraspinal and intracerebral locations are described (1–5, 8). The presence of AVMs/AVFs can cause hypertrophy of the underlying bone and tissue, resulting in limb overgrowth. This phenotype of CM-AVM is named Parkes Weber syndrome (PWS) (2, 5).
For CM-AVM2, especially, the presence of telangiectasia and episodes of spontaneous recurrent epistaxis are characteristic symptoms. The telangiectasia can be either discrete or prominent and with or without a white halo, due to vasoconstriction. Bier spots, an unspecific phenomenon with anaemic white-spotted skin, can be observed in some patients (16).
Telangiectasia and epistaxis are both well-known symptoms in hereditary haemorrhagic telangiectasia (HHT), where AVMs/AVFs also can be present. CM-AVM2 can therefore be difficult to distinguish from HHT (11).
The most relevant differential diagnoses to CM-AVM2 are CM-AVM1, HHT, Sturge-Weber syndrome (SWS) and Klippel-Trenaunay syndrome (KTS) (10, 11, 17, 18).
MATERIALS AND METHODS
This study reports a four-generation family with CM-AVM2 together with a review of published CM-AVM2 cases and cohorts with a focus on the clinical presentation. A literature search was conducted at PubMed in the period 1 February 2021 to 1 March 2021. The following keywords were used: EPHB4 capillary malformation, CM-AVM II, CM-AVM2, capillary malformation-arteriovenous malformation.
Publications with CM-AVM2 case reports or case series with known pathogenic variants in EPHB4 were included. Exclusion criteria were non-English language. The reference lists of the included papers were examined. Six articles describing CM-AVM2 cases or cohorts were found, 2 were excluded. One paper merged CM-AVM1 and CM-AVM2 in the clinical description, hence it was not possible to extract information for CM-AVM2. The other did not include clinical characteristics. Overall the literature review included 4 articles.
RESULTS
Capillary malformation-arteriovenous malformation syndrome-2 four-generation family
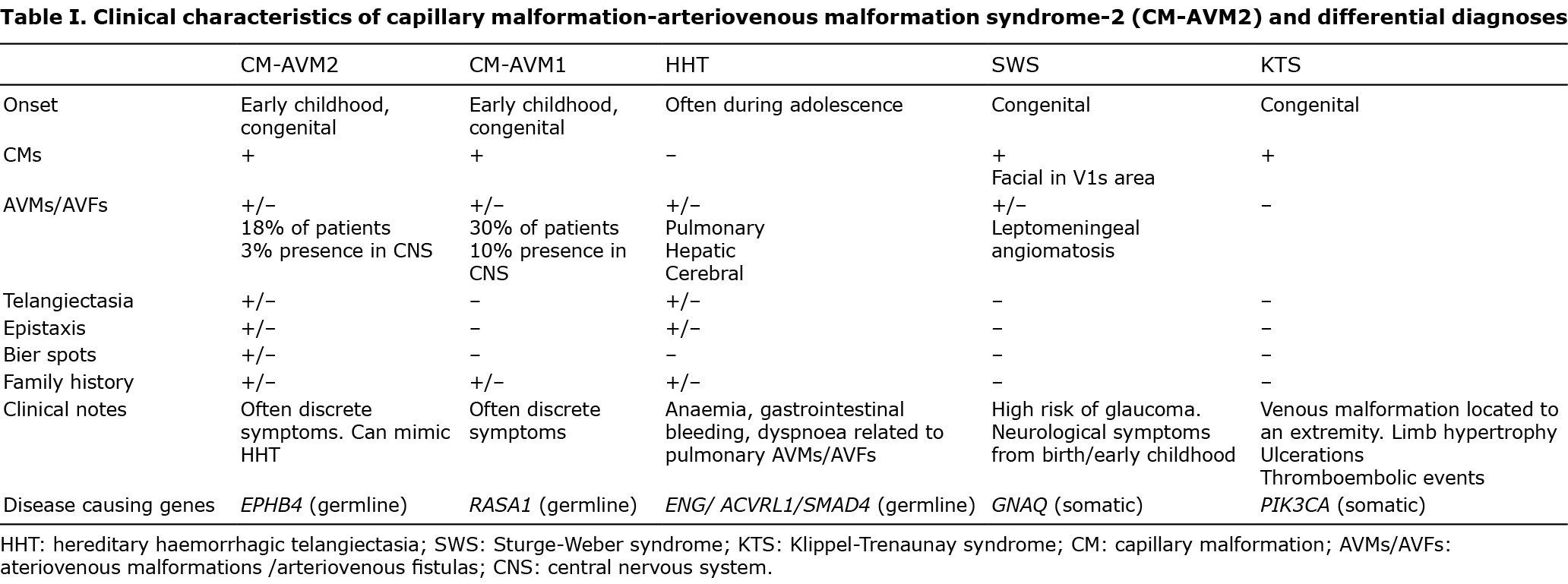
We report here a four-generation family with CM-AVM2 (Fig. 1).
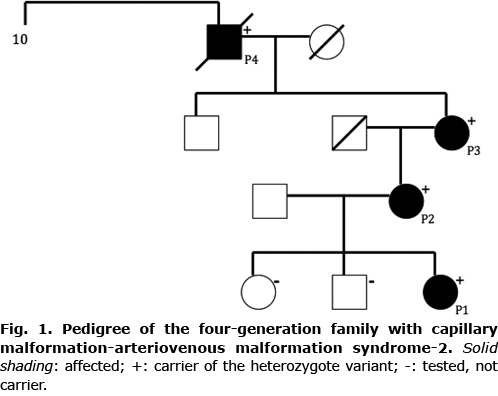
A 9-year-old girl (P1) presented with several punctate telangiectasia, mostly on her face and upper extremities, showing a halo phenomenon in many lesions. Bier spots were observed on her dorsal hands. She had a naevus araneus under the right eye and a thumbprint size macular CM on her left knee (Fig. 2) with a positive Doppler signal. She experienced episodes of recurrent epistaxis. According to her mother, perioral telangiectasias were present from birth and HHT was suspected. During her childhood the cutaneous lesions on her face and hands had gradually evolved. She was born with an infantile haemangioma at the upper back, which spontaneously resolved before the age of 2 years.
A 43-year old woman (P2), mother of P1, presented with multiple small and punctate facial telangiectasia, especially perioral, on her lips and also on her anterior neck. HHT had been suspected since birth due to intraoral telangiectasia. Some of the lesions had a white surrounding halo. She had a 2-cm macular CM over her right eyebrow with positive Doppler pulsation. Bier spots were present on both forearms. She did not report episodes of epistaxis
Cerebral magnetic resonance imaging (MRI) was performed as part of elucidation for hypothyroidism and did not reveal any cerebral AVM/AVF. In 2019 she received treatment for varicose veins on both legs and subsequently a tender prominent process was discovered in the popliteal region. A suspicion of AVM/AVF was raised, but an MRI scan did not confirm the suspicion.
A 69-year-old woman (P3), mother of P2, presented with several facial and intraoral telangiectasia and a coin-size CM in her right palm. Prominent Bier spots were seen on both forearms. Recurrent epistaxis was reported, especially during childhood and adolescence. She had been diagnosed with HHT during her pregnancy in 1976 because of epistaxis and widespread vascular skin lesions. Later in life, an HHT expert could not confirm the HHT suspicion clinically.
A 94-year-old male (P4), father of P3, had appropriate skin for his age and no CMs, telangiectasia or Bier spots. He had a prominent round tumour in his palate without palpable pulsation. He could recall earlier surgery, and remembered that the surgeons described a connection of tumour vessels to the brain. The tumour vessels had been clamped at a young age. He described episodes of epistaxis in his youth, and earlier presence of telangiectasia. HHT had been suspected for many years. However he did not fulfil diagnostic criteria, when he was examined at the national HHT centre in 2011. He had a recent episode with suspected gastrointestinal bleeding and anaemia, for which he declined further investigation.
The suspicion of HHT had been present in this family, and P3 and P4 had in fact earlier been diagnosed with HHT, but did not fulfil the diagnostic criteria for HHT, when examined by an HHT expert. Because of the presence of clinical symptoms in 4 generations and reevaluation raised the suspicion of CM-AVM, P2 had genetic analysis performed for HHT (ENG, ACVRL1, SMAD4) and CM-AVM (RASA1, EPHB4), which revealed a heterozygous missense variant in EPHB4 (NM_004444.5 (EPHB4): c.2224G>C, p.(Ala742Pro)). This variant was later confirmed in P1, P3 and P4. The 2 healthy siblings of P1 had no cutaneous manifestations and did not carry the EPHB4 variant. The variant, which had not been described in the literature or databases (ClinVar/LOVD/gnomAD), included a highly preserved nucleotide and amino acid. In silico analysis predicted that the variant was likely pathogenic (4/4 prediction software). Overall, the variant is considered likely to be pathogenic based on the above information and the segregation in this family.
Clinical data regarding capillary malformation-arteriovenous malformation syndrome-2 from the literature
A total of 127 patients were identified by a literature search, and the results are summarized in Table II. The presence of one or more CMs was reported in 114 (89.76%) of patients, while 12 (9.44%) patients presented with only 1 CM. AVMs/AVFs were present in 23 (18.1%) patients. Hence, most patients with CM-AVM2 do not have these complicating features. Localization in the central nervous system (CNS) was identified in 5 (3.9%) patients. Three of these were vein of Galen aneurysmal malformations (VGAMs) and 2 were spinal AVMs. The localization of the other AVM/AVF were face and extremities. Telangiectasia was observed in 28 patients (22%) and epistaxis was not reported sufficiently in all included papers. Bier spots were present in 20 patients (15.7%). There was not sufficient description to establish age at diagnosis, or mean age of development of cutaneous lesions.

DISCUSSION
We report here a four-generation family with a novel variant in EPHB4, which is considered likely to be pathogenic. The family was initially suspected with HHT, and later on CM-AVM and genetic testing revealed that they had CM-AVM2, which emphasizes the phenotypic overlap between these two genetic disorders.
This literature study included all published clinical data on CM-AVM2 patients with verified pathogenic variants in EPHB4. In general, the current observations confirmed the clinical findings of Amyere et al. (2), which was unsurprising, as their study comprised 110 out of 127 (86.6%) patients in the current review. The patients reported by Amyere et al. were recruited from a database of patients with cutaneous vascular malformations collected over many years. They investigated 365 patients with sporadic or familial CMs with or without fast-flow malformations, and found a pathogenic variant in EPHB4 in 54 patients, and later 56 relatives.
The literature review found that 114/127 (89%) of patients had 1 or more CMs.
23/127 (18%) of patients had a fast-flow lesion. In 5 of these (3.9%) the location was in CNS: 3 VGAMs and 2 spinal AVMs. It is notable that, in a study by Wooderchak-Donahue et al. (11), 20% (2/10) of patients had an AVM/AVF in the CNS. The current family and the 10 cases from Wooderchak-Donahue et al. (11) were initially suspected to have HHT, which might explain the high proportion of telangiectasia reported.
Epistaxis was reported in at least 9 CM-AVM2 cases. Amyere et al. (2) reported “few recurrent episodes of epistaxis”. In the current family 3 out of 4 subjects reported episodes of epistaxis, and Wooderchak-Donnahue et al. (11) described 6/10 cases with epistaxis. This indicates that epistaxis among patients with CM-AVM2 may be more frequent than first assumed.
Bier spots were present in 20/127 (15%) of cases. This is a characteristic feature, but it can be discrete and fluctuating, and therefore might be overlooked in a clinical setting.
Early development or the congenital presence of CMs, AVMs/AVFs and telangiectasia are characteristic. In contrast, the typical HHT patient will not develop cutaneous manifestations before adulthood (19).
Hereditary benign telangiectasia (HBT) is a condition described especially in older literature as a condition with widespread telangiectasia with halo (20, 21). A genetic cause for HBT has not been reported. However as the clinical phenotype is very similar to CM-AVM2, it is reasonable to suspect that it might be the same disorder. In fact the initial dermatological consideration in the current family was HBT.
The incidence of CM-AVM1/2 is not yet known. Amyere et al. (2) estimated that the theoretic prevalence of CM-AVM1/2 might be comparable to HHT (approximately 1/5,000) based on the prevalence of truncated variants in the relevant genes. Overall CM-AVM2 and CM-AVM1 are probably highly underdiagnosed, based on the mild and discrete phenotype seen in most cases, and the misdiagnosis of some patients as having HHT.
There are currently no international diagnostic criteria for CM-AVM1 or CM-AVM2. The diagnosis is suspected based on clinical signs and then verified with molecular genetic analysis. This procedure is challenged in areas where molecular genetic analyses are not accessible. CM-AVM2 would benefit from clear diagnostic criteria. Orme et al. (4) proposed diagnostic clinical criteria for CM-AVM1, based on the presence of the number of atypical CMs (3 or more) or genetic test result, or the presence of CMs and AVMs and family history. However, at that time-point CM-AVM2 had not yet been described. We suggest including telangiectasia and Bier spots or white haloes as well as early development of cutaneous lesions as a part of the diagnostic criteria. With 2 types and overlapping clinical characteristics we suggest that diagnostic criteria should be defined for CM-AVM1 and CM-AVM2 separately.
In conclusion, CM-AVM2 with heterozygous pathogenic variants in the EPHB4 gene is characterized by a complex clinical spectrum that may mimic different vascular disorders, especially HHT. More knowledge about the clinical characteristics of this condition is needed in order to establish clinical diagnostic criteria and guidelines for treatment.
The authors have no conflicts of interest to declare
REFERENCES
- Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe, S et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 2003; 73: 1240–1249.
- Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation 2017; 136: 1037–1048.
- Revencu N, Boon LM, Mendola A, Cordisco MR, Dubois J, Clapuyt P, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat 2013; 34: 1632–1641.
- Orme CM, Boyden LM, Choate KA, Antaya RJ, King BA. Capillary malformation-arteriovenous malformation syndrome: review of the literature, proposed diagnostic criteria, and recommendations for management. Pediatr Dermatol 2013; 30: 409–415.
- Revencu N, Boon LM, Mulliken JB, Enjolras O, Cordisco MR, Burrows PE, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat 2008; 29: 959–965.
- Lapinski PE, Doosti A, Salato V, North P, Burrows PE, King PD. Somatic second hit mutation of RASA1 in vascular endothelial cells in capillary malformation-arteriovenous malformation. Eur J Med Genet 2018; 61: 11–16.
- Amyere M, Aerts V, Brouillard P, McIntyre BA, Duhoux FP, Wassef M, et al. Somatic uniparental isodisomy explains multifocality of glomuvenous malformations. Am J Hum Genet 2013; 92: 188–196.
- Valdivielso-Ramos M, Martin-Santiago A, Azaña JM Hernández-Nuñez A, Vera A, Perez B, et al. Capillary malformation-arteriovenous malformation syndrome: a multicentre study. Clin Exp Dermatol 2021; 46: 300–305.
- Maruani A, Durieux-Verde M, Mazereeuw-Hautier J, Boccara O, Martin L, Chiaverini C, et al. Search for RASA1 variants in capillary malformations of the legs in 113 children: results from the French National Paediatric Cohort CONAPE. Acta Derm Venereol 2018; 98: 251–255.
- Cen Q, Sun Y, Zeng X, Liu Y, Liu F, Chen H, et al. Unilateral and segmental distribution of facial erythema: is it a real port-wine stain? Hereditas 2020; 157: 27.
- Wooderchak-Donahue WL, Akay G, Whitehead K, Briggs E, Stenvenson DA, O’Fallon B, et al. Phenotype of CM-AVM2 caused by variants in EPHB4: how much overlap with hereditary hemorrhagic telangiectasia (HHT)? Genet Med 2019; 21: 2007–2014.
- Yedidi RS, Raffi J, Sugarman J. Broadly distributed vascular macules in a pediatric patient. Cutis 2020; 105: E7–E9.
- Gourier G, Audebert-Bellanger S, Vourc’h P, Fraitag S, L’Hérondelle K, Labouche A, et al. Multiple capillary malformations of progressive onset: Capillary malformation-arteriovenous malformation syndrome (CM-AVM). Ann Dermatol Venereol 2018; 145: 486–491.
- Kim C, Ko CJ, Baker KE, Antaya RJ. Histopathologic and ultrasound characteristics of cutaneous capillary malformations in a patient with capillary malformation-arteriovenous malformation syndrome. Pediatr Dermatol 2015; 32: 128–131.
- Rodríguez Bandera AI, Feito Rodríguez M, Chiloeches Fernández C, Stewart N, Valdivielso-Ramos M. Role of colour-Doppler high-frequency ultrasonography in capillary malformation-arteriovenous malformation syndrome: a case series. Australas J Dermatol 2020; 61: 349–352.
- Kluger N, Bessis D. Bier’s spots. J Eur Acad Dermatol Venereol 2019; 33: e78–e79.
- El Hajjam M, Mekki A, Palmyre A, Eyries M, Soubrier F, Bourgault Villada I, et al. RASA1 phenotype overlaps with hereditary haemorrhagic telangiectasia: two case reports. J Med Genet 2021; 58: 645–647.
- Yu J, Streicher JL, Medne L, Krantz ID, Yan AC. EPHB4 Mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol 2017; 34: e227–e230.
- Rimmer J, Lund VJ. Hereditary haemorrhagic telangiectasia. Rhinology 2015; 53: 195–203.
- Molho-Pessach V, Agha Z, Libster D, Lere I, Burger A, Jaber S, et al. Evidence for clinical and genetic heterogeneity in hereditary benign telangiectasia. J Am Acad Dermatol 2007; 57: 814–818.
- Ujiie H, Kodama K, Akiyama M, Shimizu H. Hereditary benign telangiectasia: two families with punctate telangiectasias surrounded by anemic halos. Arch Dermatol 2010; 146: 98–99.