ORIGINAL REPORT
Clinical Features of Paediatric Inflammatory Epidermolysis Bullosa Acquisita: A Case Series Study
Yuexin ZHANG1–3#, Jingyang DANG1–3#, Ruoyu LI1–3, Xixue CHEN1–3, Xuejun ZHU1–3 and Mingyue WANG1–3
1Department of Dermatology, Peking University First Hospital, 2National Clinical Research Center for Skin and Immune Diseases and 3Beijing Key Laboratory of Molecular Diagnosis on Dermatoses, Beijing, China
#These authors contributed equally to this work.
Epidermolysis bullosa acquisita (EBA) rarely develops in childhood. This study retrospectively recruited paediatric patients with EBA (age ≤ 16 years), diagnosed by clinical and histopathological features and results of immunofluorescence, immunoblotting and enzyme-linked immunosorbent assay (ELISA), and reviews their clinical manifestations, histopathology, immunological features, and responses to various treatments. All 7 included patients presented with inflammatory EBA. Among them, 3 had a bullous pemphigoid-like phenotype. Pathologically, in addition to dermal–epidermal blistering, in all patients, the distribution of neutrophils was superficial perivascular or interstitial, or in the dermal papilla. Mixed neutrophils and eosinophils were detected in 2 of the 3 patients with bullous pemphigoid-like phenotypes. In addition to treatment with glucocorticoids, dapsone was administered in 4 patients, while thalidomide and sulfasalazine were administered in 1 patient. All patients responded to the these therapies. Relapse was mainly due to reduction and cessation of glucocorticoids. In conclusion, EBA in childhood may be unique, and thus distinct from its adult counterpart. Specific treatment and follow-up protocols are required for therapy of this rare autoimmune skin disease in children.
SIGNIFICANCE
Epidermolysis bullosa acquisita in childhood appears to be different from in adults. In our study, the participants spanned an age range of 3 to 16 years, comprising 3 male and 4 female individuals. Children usually presented with inflammatory skin lesions on flexural or intertriginous areas. The distribution of mucosal lesions could be random. Pathologically, except for subepidermal blistering, the infiltrating cells in the dermis were predominantly neutrophils. In the study cohort paediatric epidermolysis bullosa acquisita was less comorbid with other disorders compared to Western countries, such as inflammatory bowel disease and thyroiditis. Relapse was mainly due to reduction and cessation of glucocorticoids.
Citation: Acta Derm Venereol 2024; 104: adv11917. DOI https://doi.org/10.2340/actadv.v104.11917.
Copyright: © Published by Medical Journals Sweden, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/)
Submitted: Mar 23, 2023; Accepted: Nov 8, 2023; Published: Jan 25, 2024
Corr: Mingyue Wang, Department of Dermatology, Peking University First Hospital, Beijing 100034, China. E-mail: wangmy@pku.edu.cn
Competing interests and funding: The authors have no conflicts of interest to declare.
INTRODUCTION
Epidermolysis bullosa acquisita (EBA) is a rare, acquired, chronic autoimmune bullous disease (AIBD) characterized by autoantibodies (mainly immunoglobulin G (IgG) class) targeting type VII collagen (COL7) (1). EBA was most likely first reported by Elliot in 1895 as a non-hereditary epidermolysis bullosa after excluding other known bullous diseases (2). The term “epidermolysis bullosa acquisita” was first proposed by Kablitz (3). In 1971, Roenigk et al. (4) established the first diagnostic criteria for EBA. However, the incidence and prevalence of EBA remain unknown and appear to be different in different populations. An incidence rate between 0.2 and 1.1 new cases per million/year in Singapore, Kuwait, France and Germany has been reported (5, 6). Hübner et al. (7) reported that the prevalence of EBA among individuals under 18 years old is 1.2 patients per million children in Germany. A meta-analysis revealed that EBA affects all age groups (median 50 years, range 1–94 years) with an equal sex distribution (8). EBA has sometimes been associated with other chronic inflammatory diseases, such as Crohn’s disease, other AIBDs, thyroiditis, immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, coeliac disease, alopecia areata, hepatitis C virus infection, and rheumatoid arthritis (9, 10).
Clinically, EBA is subdivided into an inflammatory and a non-inflammatory phenotype (11). Non-inflammatory EBA causes blisters and skin fragilities in trauma-prone positions, such as acral areas and the extensor skin surface. This phenotype is characterized by dystrophic changes, including milia and atrophic scars, pigmentary changes, loss of nails, digital contractures, and oesophageal strictures. In the inflammatory forms, blisters occur in both non-traumatic and trauma-prone areas, which generally heal with minimal scarring and milia formation. The inflammatory form can mimic almost all other chronic bullous diseases, and its clinical differentiation from bullous pemphigoid (BP), Brunsting-Perry pemphigoid, and linear immunoglobulin A (IgA) bullous dermatosis (LABD) may be difficult (11, 12). However, the manifestation of the disease may change with its course, and the characteristics of the 2 aforementioned phenotypes may coexist in the same patient (mixed phenotype). No significant differences between non-inflammatory and BP-like EBA were found previously, in terms of sex, age of onset, oral involvement, enzyme-linked immunosorbent assay (ELISA) titres, or time to remission (6, 13). Inflammatory EBA is more common in children, and more often presents like other inflammatory blistering diseases, with more frequent mucosal involvement than the classic non-inflammatory EBA frequently seen in adults. Childhood EBA seems to have a more favourable prognosis and course compared with its adult counterpart, both in rates of remission and in terms of control with pharmacotherapy (14).
The aims of this study are to analyse the clinical presentation, pathological manifestations, immunological features, responsiveness to treatment, and prognosis of 7 patients diagnosed with EBA at the Department of Dermatology of Peking University First Hospital, Beijing, China.
MATERIALS AND METHODS
Study design and subjects
All data were collected retrospectively from the medical records of non-adult patients (age ≤ 16 years) diagnosed with EBA over a 17-year period (January 1, 2003–January 1, 2021) at the Department of Dermatology of Peking University First Hospital, Beijing, China, also known as the National Clinical Research Center for Skin and Immune diseases, which is a referral centre for rare diseases. The EBA diagnosis was confirmed by clinical, histological features, immunofluorescence, immunoblot analyses, and ELISA in compliance with the diagnostic criteria for EBA developed by the International Bullous Diseases Group (11). EBA was clinically classified into the following phenotypes: non-inflammatory, inflammatory (BP-like, LABD-like, and Brunsting-Perry-like pheno-types), or mixed (11). In the S3 guideline for mucous membrane pemphigoid (MMP) (15, 16), there is a widely accepted consensus that the specific autoantibody reactivity and immunoglobulin class profile should not be considered or specified when determining the terminology and classification of MMP. Consequently, patients with predominant mucosal lesions are now diagnosed with MMP, regardless of the targeted antigens. Patients previously classified as having MMP-like EBA are now categorized as MMP. Therefore, this paper does not include MMP-like EBA as one of the phenotypes of EBA. Patients were directly classified as having inflammatory EBA without further classification if inflammatory EBA phenotypes had no characteristics of BP or Brunsting-Perry pemphigoid, or the immunofluorescence (IF) for IgA antibody was negative. All patients included in this study provided written informed consent.
Data collection
Demographic and clinical data were retrospectively recorded from clinical charts completed at the initial visit and at each subsequent follow-up visit. Data on rash and oral lesions were obtained from clinical photographs taken at the time of the patients’ first visit.
Biopsy was followed by direct immunofluorescence (DIF) assay for IgG, IgA, IgM, and C3 of skin samples. Indirect immunofluore-scence (IIF) analyses of normal human skin and 1 M NaCl-split-human skin (ss-IIF) for both IgG and IgA antibodies were then conducted. Immunoblotting was carried out using normal human epidermal extract to identify the presence of autoantibodies against desmoglein (Dsg)1, Dsg3, BP180, BP230, envoplakin, and periplakin in the patient’s serum. Immunoblotting analysis was performed to detect antibodies against COL7 and laminin γ1 using normal human dermal extracts. Furthermore, ELISAs of the recombinant non-collagenous 16A domain of BP180 (cut-off values: IgG < index 20) (Euroimmun, Lűbeck, Germany), BP230 C-terminal domains (cut-off values: IgG < index 20) (Euroimmun), COL7 non-collagenous (NC) 1 and NC2 domains (cut-off values: IgG < index 6.14) (MBL, Nagoya, Japan) were performed. We analyzed the suspected cases using the immunoblot strips kindly provided by Dr. Lars Komorowski at Euroimmun, Germany, which contained COL7.
The observation endpoints for EBA were defined by modifying the terms for BP recommended by an international panel of experts (17). Complete remission off therapy (CR) was defined as the absence of new or established lesions for at least 2 months without treatment. Complete remission on minimal therapy (CRT) referred to the absence of new or established lesions while receiv-ing minimal therapy for at least 2 months. Partial remission off therapy (PR) described transient new lesions that healed within 1 week without treatment for at least 2 months. Partial remission on minimal therapy (PRT) was defined as the presence of transient new lesions that healed within 1 week while receiving minimal therapy for at least 2 months.
RESULTS
Screening for paediatric patients with epidermolysis bullosa acquisita
Seven patients diagnosed with EBA were enrolled in the study. It is notable that these patients showed no evidence of systemic lupus erythematosus.
Baseline characteristics
Median age at disease onset was 10.7 (range 3–16) years, with 3 (3/7) male and 4 (5/7) female patients. All children presented with inflammatory EBA, 3 of whom had a BP-like phenotype (Table I). Most patients had no apparent cause. One patient (patient 3) had received an anti-influenza vaccine 1 day before disease onset, which may be a possible cause.
Three patients had an additional diagnosis; 2 had atopic dermatitis (AD) and 1 had eczema (Table I). One patient (patient 7) had paroxysmal abdominal pain before and during the onset of EBA. Post-treatment, the rash was improved, and the abdominal pain was relieved. Nevertheless, this patient did not visit a gastroenterologist and thus had no digestive diagnosis. In addition, 2 patients with BP-like EBA had a history of AD and allergies. None of the patients had any comorbidities, such as inflammatory bowel disease (IBD), diabetes mellitus, thyroiditis, other AIBDs, rheumatoid arthritis, cryoglobulinaemia, psoriasis, cancer, or haemophilia A.
Although these patients were diagnosed with inflammatory EBA, they showed different degrees of skin fragility. The lesions in children were most likely to start at a site easily stimulated by external forces, including the back of the hands (patients 1 and 3), lower eyelid (patients 2 and 7), perioral region (patients 4 and 5), and oral cavity (patient 6). On the trunk, the flexural or intertriginous areas were often involved, such as the armpit, cleavage, navel, groin and neck (Fig. 1a–d). Regarding the extremities, the extensor surfaces were the most commonly and severely affected sites. Extremity lesions in 4 patients were predominantly located on the extensor side (Fig. 1e).
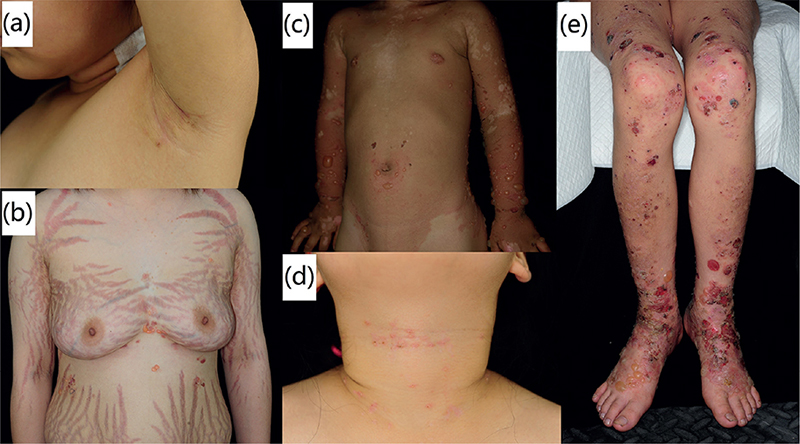
Fig. 1. Clinical manifestations of 7 paediatric cases of epidermolysis bullosa acquisita. The skin lesions are distributed in the flexural or intertriginous areas of the body, including the: (a) armpit, (b) cleavage, (c) navel, (d) neck and (e) groin. (e) Lesions are located on the extensor surfaces of the extremities. We have obtained permission from the parents for the clinical photos and laboratory data to be featured in papers, magazines, or any other works or products.
The most easily affected parts for mucosa were different. Six patients with EBA had mucosal involvement. In 2 patients (patients 2 and 3), only oral mucosal sites were affected; in 1 patient (patient 1), the vulva and anus were affected; and in 3 patients (patients 5, 6 and 7), both sites were involved. No predominance of any oral mucosal lesion on a particular area of the oral mucosa was observed.
Histopathological features and immunological findings
Subepidermal blisters were found in 6 patients (patients 1, 2 and 4–7). The other patient (patient 3) showed vacuolar alteration along the dermal–epithelial junction. In all patients, neutrophils had a superficial perivascular or interstitial distribution or such in the dermal papilla, while there were mixed neutrophils and eosinophils in 2 of 3 BP-like phenotypes (patients 2 and 5).
DIF was performed in 6 patients with EBA. Two of these patients showed IgG deposition to the basement membrane zone (BMZ), and 3 showed C3 deposition; no patients showed IgA deposition (Table II). For IIF of normal human skin, all patients showed IgG anti-BMZ antibodies (Fig. S1). In ss-IIF, all patients had IgG located at the dermal side at titres from 1:40 to over 1:160, while 1 patient showed IgA reactivity with dermal side, at a very low titre of 1:8 (Table II, Fig. 2).
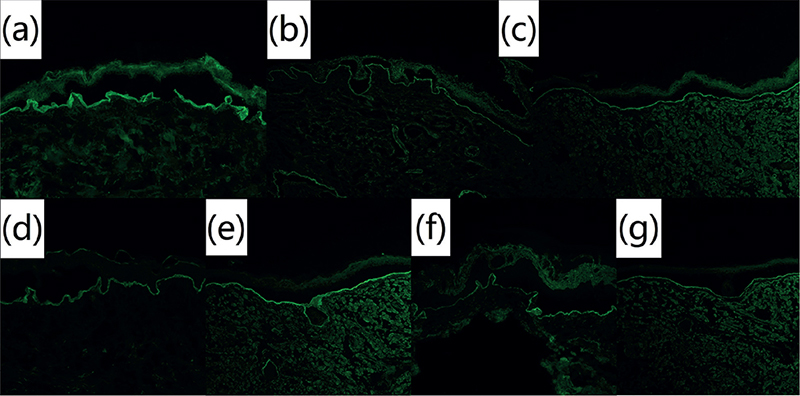
Fig. 2. Indirect immunofluorescence on salt-split skin of 7 paediatric cases of epidermolysis bullosa acquisita. Circulating immunoglobulin G (IgG) autoantibodies were found to bind to the dermal side of 1M NaCl-split normal human skin through indirect immunofluorescence at a serum dilution of 1:40. Pictures 2a-2g: patients 1-7 (original magnification, ×100).
No EBA sera reacted with epidermal autoantigens by immunoblotting (Table II). However, on the normal human dermal extracts, IgG antibodies to the 290-kDa COL7 were detected in all patients (Fig. 3). IgA antibodies were detected in 1 patient (patient 5) at a titre of 1:5. In contrast, IgG reactivity with laminin γ1 was not detectable in any of the patients using normal human dermal extracts (Fig. 3).
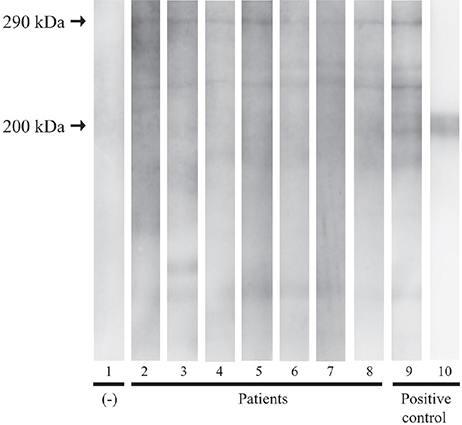
Fig. 3. Immunoblotting of normal human dermal extract. Serum from the patients (lanes 2–8) demonstrated a reaction with 290 kDa type VII collagen. Lane 1 was a negative control, while lane 9 served as a positive control with a confirmed case of epidermolysis bullosa acquisita. Lane 10 served as a control with the primary antibody against anti-laminin γ1.
Therapies and outcomes
The therapeutic regimen depended on the severity of the disease, patient-specific characteristics, and patients’ preferences (Table III). Systemic corticosteroids were administered in 5 patients. One patient did not receive systemic corticosteroids because no consent was granted by the parents. The other patient responded quite well to dapsone, with no need for administration of corticosteroids. Dapsone was used in 4 patients, and thalidomide and sulfasalazine were each used in 1 patient. All patients responded to these therapies.
At the final follow-up visit, CR was observed in 43% of individuals (3/7), while PRT was present in 14% (1/7). The times to achieve CR for patients 3, 4, and 5 were 370, 395, and 580 days, respectively. Patient 4 stopped glucocorticoid herself when the dose tapered to 2 mg/day, and the disease relapsed after 5 months. We tried to stop the glucocorticoid, only use sulfasalazine at 0.25 g/day as maintenance therapy for patient 7, who reached PRT, but the disease relapsed 1 month after stopping the steroid. The condition recurred twice during drug reduction in patient 2, who was one of the patients who did not use systemic corticosteroids. Patient 1 was lost to follow-up. Among the 4 patients with relapse, 2 cases (patients 2 and 6) were due to glucocorticoid reduction, and 2 cases (patients 4 and 7) were caused by discontinuation of the glucocorticoid. Notably, patient 4 had a history of cold before the relapse, which might be a reason for this development.
The most common complications caused by the EBA treatments applied, were obesity and Cushing’s syndrome, which were detected in 3 patients. No other side-effects of systemic corticosteroids, such as infections, gastric ulcers, and osteoporosis, were found during follow-up.
DISCUSSION
EBA is a rare autoimmune bullous disease resulting in vesicle and bullae formation on the skin and erosions on the mucous membranes. The diagnosis of paediatric inflammatory EBA can be challenging, as it shares clinical and histopathological features with other bullous diseases, such as BP and LABD. If available, histopathology, DIF, IIF with salt-split skin, immunoblotting and ELISA are useful diagnostic tests.
In our study, the participants spanned an age range of 3 to 16 years, comprising three male and four female individuals. All patients in this study cohort presented with inflammatory type EBA. Children usually presented with inflammatory skin lesions on flexural or intertriginous areas with/without mucous membrane lesions. The distribution of EBA lesions in the mucosa may be random, which is also easily overlooked. Histopathologically, except for subepidermal blistering, the infiltrating cells in the dermis were predominantly neutrophils. Paediatric EBA in the current study cohort is less comorbid with other disorders compared to Western countries, such as IBD and thyroiditis. The relapse was mainly due to the reduction and cessation of glucocorticoids. Therefore, the clinical presentations, pathological manifestations, responsiveness to treatment, and prognosis of children with EBA are different from those of adults. Specific treatment and follow-up characteristics are required for this rare autoimmune skin disease in children.
One patient (patient 3) had a clear history of influenza vaccination. To the best of our knowledge, no cases of influenza vaccine-induced EBA have been reported previously. However, several reports have been published of BP caused by vaccines (18–22), probably through immune system stimulation by an unexplained mechanism. Thus, we suspect that vaccination might have caused EBA by enhancing an autoimmune response in an immunologically predisposed individual. Treatment of this patient achieved a considerable beneficial effect; the disease was controlled with dapsone, 50 mg/day, and the drug was stopped entirely after 1 year, with no recurrence thereafter.
Only 1 patient (patient 7) had symptoms of abdominal pain. Colonoscopy or intestinal histopathological examinations were not performed due to the few complaints of related symptoms, so that subclinical involvement might have been missed. Korean researchers reported that none of the patients in their cohort had EBA with comorbid IBD (6) due to the lower prevalence of IBD in Korea and the differences in the IBD-associated genomics between Korea and Western countries (23). These reasons may explain the low incidence of IBD among EBA patients in our cohort.
Most types of infiltrating cells in the patients’ biopsies were neutrophils. The lesions in 2 of the patients who had high counts of eosinophils might have been a complex manifestation of the interaction between AD and EBA, or EBA pathophysiology itself. Tissue eosinophilia was previously found in skin lesions biopsies of AD (24). Currently, there is no evidence that EBA is associated with the pathogenesis of AD.
Treatment of EBA is difficult. Randomized therapeutic trials are lacking, and no clear optimal treatment approach has been agreed (25). Dapsone and low-dose prednisolone appear to be beneficial in childhood EBA (26). Therapeutic effects have been achieved in patients with glucocorticoid administration alone or such combined with dapsone, thalidomide, and colchicine. The reason for the relapse in the current study was mainly due to the reduction and cessation of glucocorticoids. Therefore, appropriate and regular treatment is essential. Most of the patients had 1 relapse, but the patients treated without glucocorticoids had 2 relapses. Therefore, it is necessary to administer appropriate doses of glucocorticoids.
The current study has some limitations. First, the patient sample size was relatively small; only 7 patients were included in the study. Secondly, the DIF assays were not all performed in our institute, Department of Dermatology, Peking University First Hospital, Beijing, China. In addition, the patient lacks data on serration pattern analysis, a technique first reported by Vodegel et al. for differentiation between AIBDs with anti-COL7 antibodies and other pemphigoid diseases (27). However, in order to follow precisely and completely the steps of the clinical and diagnostic criteria for EBA developed by the International Bullous Diseases Group (11), we performed IIF, ss-IIF, immunoblotting and ELISA, whose results were all consistent with the diagnosis of EBA. Therefore, we obtained sufficient evidence to diagnose EBA. Thirdly, this was a retrospective study. Further research, using a multicentric, multidisciplinary prospective approach, including a large number of patients, is necessary to improve our knowledge and management of this disease.
ACKNOWLEDGEMENTS
We would like to express our sincere gratitude for the support provided by Dr Lars Komorowski from Euroimmun in supplying the immunoblot strips. We are greatly indebted to Professor Takashi Hashimoto at Osaka City University for his kind and generous academic assistance.
REFERENCES
- Papara C, Karsten CM, Ujiie H, Schmidt E, Schmidt-Jiménez LF, Baican A, et al. The relevance of complement in pemphigoid diseases: a critical appraisal. Front Immunol 2022; 13: 973702.
- Elliott GT. Two cases of epidermolysis bullosa. J Cutan Genitourin Dis 1895; 13: 10–18.
- Kablitz R. Ein Beitrag Zur Frage der Epidermolysis bullosa (heriditaria et acquisita). These Inaug Rostock Diss 1904.
- Roenigk HHJ, Ryan JG, Bergfeld WF. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Arch Dermatol 1971; 103: 1–10.
- Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol 2017; 13: 157–169.
- Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol 2011; 91: 307–312.
- Hübner F, König IR, Holtsche MM, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases among paediatric patients in Germany. J Eur Acad Dermatol Venereol 2020; 34: 2600–2605.
- Iwata H, Vorobyev A, Koga H, Recke A, Zillikens D, Prost-Squarcioni C, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis 2018; 13: 153.
- Guerra L, Pacifico V, Calabresi V, De Luca N, Castiglia D, Angelo C, et al. Childhood epidermolysis bullosa acquisita during squaric acid dibutyl ester immunotherapy for alopecia areata. Br J Dermatol 2017; 176: 491–494.
- Iranzo P, Herrero-González JE, Mascaró-Galy JM, Suárez-Fernández R, España A. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol 2014; 171: 1022–1030.
- Prost-Squarcioni C, Caux F, Schmidt E, Jonkman MF, Vassileva S, Kim SC, et al. International Bullous Diseases Group: consensus on diagnostic criteria for epidermolysis bullosa acquisita. Br J Dermatol 2018; 179: 30–41.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol 2013; 2013: 812029.
- Kim JH, Kim YH, Kim S, Noh EB, Kim SE, Vorobyev A, et al. Serum levels of anti-type VII collagen antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol 2013; 27: e224–230.
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita: a review. Pediatr Dermatol 2021; 38: 1047–1050.
- Rashid H, Lamberts A, Borradori L, Alberti-Violetti S, Barry RJ, Caproni M, et al. European guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European Academy of Dermatology and Venereology – Part I. J Eur Acad Dermatol Venereol 2021; 35: 1750–1764.
- Schmidt E, Rashid H, Marzano AV, Lamberts A, Di Zenzo G, Diercks GFH, et al. European Guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European Academy of Dermatology and Venereology – Part II. J Eur Acad Dermatol Venereol 2021; 35: 1926–1948.
- Murrell DF, Daniel BS, Joly P, Borradori L, Amagai M, Hashimoto T, et al. Definitions and outcome measures for bullous pemphigoid: recommendations by an international panel of experts. J Am Acad Dermatol 2012; 66: 479–485.
- Baykal C, Okan G, Sarica R. Childhood bullous pemphigoid developed after the first vaccination. J Am Acad Dermatol 2001; 44: 348–350.
- Oranje AP, Vuzevski VD, van Joost T, ten Kate F, Naafs B. Bullous pemphigoid in children. Report of three cases with special emphasis on therapy. Int J Dermatol 1991; 30: 339–342.
- Cambazard F, Thivolet J, Mironneau P. Bullous pemphigoid in a 4-month-old boy. Br J Dermatol 1994; 131: 449–451.
- Bisherwal K, Pandhi D, Singal A, Sharma S. Infantile bullous pemphigoid following vaccination. Indian Pediatr 2016; 53: 425–426.
- Guerra L, Pedicelli C, Fania L, De Luca N, Condorelli AG, Mazzanti C, et al. Infantile bullous pemphigoid following vaccination. Eur J Dermatol 2018; 28: 708–710.
- Kim ES, Kim WH. Inflammatory bowel disease in Korea: epidemiological, genomic, clinical, and therapeutic characteristics. Gut Liver 2010; 4: 1–14.
- Kiehl P, Falkenberg K, Vogelbruch M, Kapp A. Tissue eosinophilia in acute and chronic atopic dermatitis: a morphometric approach using quantitative image analysis of immunostaining. Br J Dermatol 2001; 145: 720–729.
- Kirtschig G, Murrell D, Wojnarowska F, Khumalo N. Interventions for mucous membrane pemphigoid and epidermolysis bullosa acquisita. Cochrane Database Syst Rev 2003; 2003: Cd004056.
- Mayuzumi M, Akiyama M, Nishie W, Ukae S, Abe M, Sawamura D, et al. Childhood epidermolysis bullosa acquisita with autoantibodies against the noncollagenous 1 and 2 domains of type VII collagen: case report and review of the literature. Br J Dermatol 2006; 155: 1048–1052.
- Arora S, Shetty VM, Rao CR, Pai SB, Rao R. Serration pattern analysis as a practical adjunct tool for categorization of subepidermal autoimmune blistering diseases. Indian J Dermatol Venereol Leprol 2021; 87: 778–786.