SHORT COMMUNICATION
A Rare Case of Anetodermic Mycosis Fungoides
Brooke KENNAMER1#, Dilshad SACHEDINA1#, Neha NAGRANI2, Candice BREM2 and Debjani SAHNI1
1Department of Dermatology, Boston Medical Center, 609 Albany Street, Boston, MA 02118 USA and 2Section of Dermatopathology, Department of Dermatology, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, USA. E-mail: dsachedina@gmail.com
#Joint-first author.
Citation: Acta Derm Venereol 2023; 103: adv11926. DOI https://doi.org/10.2340/actadv.v103.11926.
Copyright: © Published by Medical Journals Sweden, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/)
Accepted: Jun 11, 2023; Published: Jul 3, 2023
INTRODUCTION
Mycosis fungoides (MF) accounts for approximately 50% of primary cutaneous T-cell lymphomas (1, 2). Anetoderma arising secondary to MF is extremely rare, with only 3 reported cases in the literature to date (3, 4). Anetoderma is a cutaneous disorder characterized by loss of dermal elastic tissue resulting in oval atrophic papules or macules that are easily depressible or protrude outwards due to herniation of underlying subcutaneous tissue. Anetoderma may be the primary presentation or it may be a secondary phenomenon due to a multitude of other dermatoses (5).
While anetodermic MF is not included in the 2018 World Health Organization – European Organization for Research and Treatment of Cancer Classification (WHO-EORTC) for primary cutaneous lymphomas (2), it has been reported as a clinicopathological variant (5). We report here a rare case of anetodermic MF, both to increase the clinical and histopathological awareness of this unusual variant of MF and to demonstrate response to treatment.
CASE REPORT
A 37-year-old Caucasian male presented with a 5-year history of a progressive, scaly, itchy rash on his trunk and extremities. An outside biopsy, obtained several years previously, was non-diagnostic. On examination, the patient had numerous erythematous patches and indurated plaques with slight scale, covering approximately 40% of the patient’s body surface area. In addition, there was alopecia and scattered soft, compressible skin-coloured to pink papules, largely limited to the patches and plaques (Fig. 1). An incisional biopsy from 1 of the plaques revealed changes consistent with cutaneous T-cell lymphoma. An additional punch biopsy from 1 of the soft papules within a plaque revealed an epidermal collarette with epidermotropism, folliculotropism, syringotropism and a dense underlying atypical lymphocytic infiltrate (Fig. 2).
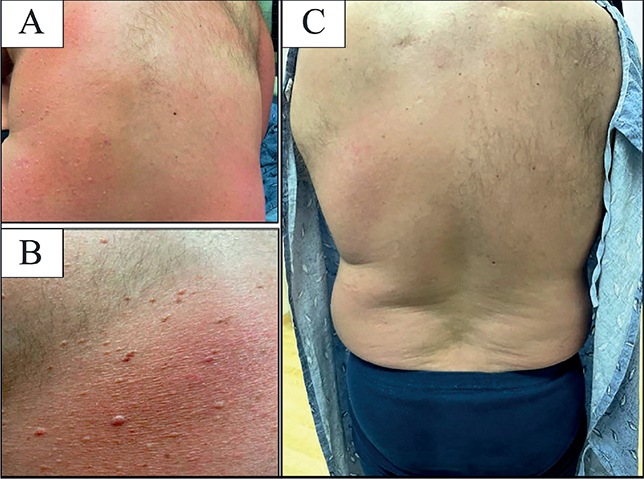
Fig. 1. Clinical photographs. (A, B) Skin on the patient’s back prior to treatment with bexarotene, demonstrating patches, plaques and scattered soft skin-coloured to pink papules. (C) Skin following treatment.
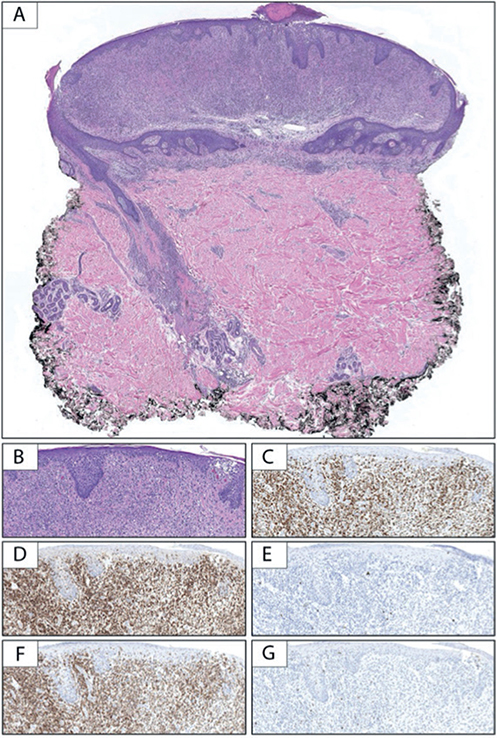
Fig. 2. Pathology of lesions. (A) Low-power examination of the punch biopsy demonstrates an epidermal collarette with a dense predominantly dermal lymphocytic infiltrate extending along appendageal structures (haematoxylin and eosin; H&E, 10×). (B) Higher power examination additionally reveals epidermotropism of atypical lymphocytes (H&E, 100×). The lymphocytic infiltrate is composed of (C) CD3 (+) T-cells with (D) CD4 predominating over (E) CD8. While there is preservation of (F) CD5, there is significant loss/absence of (G) CD7 (B–G, 100×).
Immunohistochemical staining revealed the infiltrate to be composed of CD3 (+), CD5 (+) T-cells, with CD4 predominating over CD8 and loss of CD7 consistent with MF. Given the unusual clinical presentation and the architecture of the specimen with the presence of an epidermal collarette, an elastic stain was performed, revealing near-complete absence of elastic fibres in the superficial to mid-dermis surrounding the dense lymphocytic infiltrate (Fig. 3). Taken together, these findings were consistent with folliculotropic and syringotropic MF with secondary anetoderma. There was no palpable lymphadenopathy, and blood Sézary flow cytometry ruled out any blood involvement by the disease.
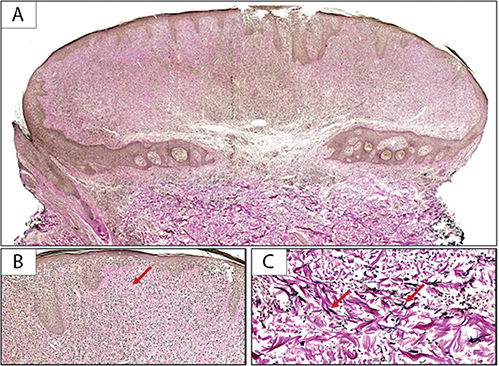
Fig. 3. (A, B) Elastic tissue stain shows near-complete absence of elastic fibres (red arrow) in the lesion (Verhoeff Van Gieson stain; (A) 10×, (B) 100×) in comparison with the preserved (C) elastic fibres (red arrows) in the underlying uninvolved dermis (Verhoeff Van Gieson; 200×).
Given the disease extent and the dense nature of the infiltrate, skin-directed treatment alone was considered unlikely to be successful. Therefore, the patient was advised to start oral bexarotene together with narrowband UVB phototherapy (nbUVB). He developed iatrogenic hypertriglyceridaemia, which stabilized on treatment with 160 mg/day fenofibrate and omega-3 fatty acids 3 g/day. After 3 months of nbUVB and up-titration of the oral bexarotene to 500 mg/day, the patient’s erythema started to improve, and the papules flattened. At various stages of his treatment course, the patient was on either bexarotene or nbUVB due to outside factors. It was notable that his disease responded best when he was taking both treatments simultaneously. His disease had improved significantly at his 5-month follow-up, as seen in Fig. 1c. To date, he has been followed up for 20 months, and is well controlled symptomatically on a combination of bexarotene and nbUVB.
DISCUSSION
Classic MF can progress from patches to plaques, and ultimately to tumours (6). Currently, the WHO-EORTC classification for primary cutaneous lymphomas recognizes only 3 clinically relevant subtypes of MF: folliculotropic MF, pagetoid reticulosis, and granulomatous slack skin. However, several other clinicopathological variants of MF exist, including anetodermic MF (6), which often present in the setting of classic MF (1) with unclear clinical and prognostic implication. Anetodermic MF appears to be a particularly rare, or possibly under-recognized, clinicopathological variant of MF as, with the inclusion of this case, only 4 cases have been reported in the literature in English to date (6).
Anetoderma is a condition characterized histologically by loss of dermal elastic tissue (7). It can be primary with no underlying disease process, where lesions occur on otherwise normal appearing skin. More commonly it is secondary and occurs in the setting of another primary dermatosis with atrophic papules in a background of the underlying condition. Secondary anetoderma has also been rarely reported in association with a handful of primary cutaneous B-cell lymphomas (8, 9).
The histology of anetodermic MF is similar to classic MF, with the additional feature of loss of dermal elastic fibres, as demonstrated by elastic tissue stains (orcein and/or Verhoeff–Van Gieson) (3, 5). The mechanism for the destruction of the elastic fibres remains unclear. Requena et al. (3) hypothesized that the dermal T-helper lymphocytes release a substance resulting in fragmentation and distortion of elastic fibres that are subsequently enveloped by macrophages. Ghomrasseni et al. (5) further postulated that anetoderma results from an imbalance of metalloproteinases and inhibitors of metalloproteinases, leading to loss of elastic tissue.
Granulomatous slack skin (GSS), another variant of MF, is the primary alternative diagnosis to anetodermic MF. The areas involved are different between the conditions, with GSS tending to favour the axillae and inguinal folds. Histopathology for GSS shows a diffuse granulomatous infiltrate, which involves the entire dermis and plenty of multinucleated giant cells. These multinucleated giant cells often digest distorted elastic fibres, leading to elastophagocytosis. On the other hand, anetodermic MF shows a lymphocyte-rich infiltrate with few multinucleate giant cells and less frequent elastophagocytosis (3).
Requena et al. (3) reported the first 2 patients with anetodermic MF, who presented with soft exophytic nodular lesions arising within plaques of MF. The third case of anetodermic MF was described by Amitay-Laish et al. (4) who reported on 20 patients with unilesional MF, all of whom showed folliculotropism on skin biopsy, with 1 of the patients (an adult male) also demonstrating anetoderma. The full demographic details of the patient were not disclosed in the article, nor was it described how long the erythematous patch and anetodermic macules were present; therefore, due to lack of follow-up data, we cannot comment on the prognostic significance. Although these are just a handful of cases, from which we cannot draw definitive conclusions, it is of interest that all 4 cases, including the current case, are adult males.
Regardless, the 3 patients described in the literature with secondary anetoderma were treated with diverse treatment modalities given their unique clinical situations (3, 4). One patient received systemic therapy with interferon alpha-2a and interleukin (IL)-12, but his disease course was complicated by a second unrelated malignancy, resulting in death (3). Another was treated with imiquimod 5% cream 3 times weekly, but follow-up was not discussed (3) and the third case with unilesional anetodermic MF received radiotherapy (4). While most of the MF plaque improved with treatment, the anetodermic macules persisted and became more prominent in appearance post-radiation. Resolution of the anetoderma itself is not mentioned in any of the known cases in the literature.
Anetodermic MF is a rare variant of MF, which may be underdiagnosed due to the lack of awareness of this clinicopathological variant and limited documentation in the literature. Although several hypotheses have been put forward, the underlying mechanism leading to the destruction of elastic fibres in anetodermic MF is not known and requires further investigation. Further study and documentation of anetodermic MF may elucidate its prognosis and response to treatment.
REFERENCES
- Song SX, Willemze R, Swerdlow SH, Kinney MC, Said JW. Mycosis fungoides: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol 2013; 139: 466–490.
- Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019; 133: 1703–1714.
- Requena L, González-Guerra E, Angulo J, DeVore AE, Sangueza OP. Anetodermic mycosis fungoides: a new clinicopathological variant of mycosis fungoides. Br J Dermatol 2008; 158: 157–162.
- Amitay-Laish I, Feinmesser M, Ben-Amitai D, Fenig E, Sorin D, Hodak E. Unilesional folliculotropic mycosis fungoides: a unique variant of cutaneous lymphoma. J Eur Acad Dermatol Venereol 2016; 30: 25–29.
- Ghomrasseni S, Dridi M, Gogly B, Bonnefoix M, Vabres P, Venencie PY, et al. Anetoderma: an altered balance between metalloproteinases and tissue inhibitors of metalloproteinases. Am J Dermatopathol 2002; 24: 118–129.
- Muñoz-González H, Molina-Ruiz AM, Requena L. Clinicopathologic variants of mycosis fungoides. Actas Dermosifiliogr 2017; 108: 192–208.
- Lewis KG, Bercovitch L, Dill SW, Robinson-Bostom L. Acquired disorders of elastic tissue: Part II. decreased elastic tissue. J Am Acad Dermatol 2004; 51: 165–185; quiz 186–168.
- Cerroni L. Skin lymphoma: the illustrated guide. 5th edn. Hoboken, NJ: Wiley-Blackwell; 2020.
- Zattra E, Pigozzi B, Bordignon M, Marino F, Chiarion-Sileni V, Alaibac M. Anetoderma in cutaneous marginal-zone B-cell lymphoma. Clin Exp Dermatol 2009; 34: e945–948.