ORIGINAL REPORT
Incidence and Prevalence of 73 Different Genodermatoses: A Nationwide Study in Sweden
Theofanis ZAGORAS1,2*, Rahime INCI1,3*, Despoina KANTERE1,3, Peter HOLMSTRÖM1, Jenny BROSTRÖM1, Martin GILLSTEDT1,3, Sam POLESIE1,3 and Sirkku PELTONEN1,3–5
1Department of Dermatology and Venereology, Institute of Clinical Sciences, Sahlgrenska Academy, University of Gothenburg, 2Region Västra Götaland, Sahlgrenska University Hospital, Department of Clinical Genetics and Genomics, 3Region Västra Götaland, Sahlgrenska University Hospital, Department of Dermatology and Venereology, Gothenburg, Sweden, 4Department of Dermatology and Allergology, University of Helsinki and 5Skin and Allergy Hospital, Helsinki University Hospital, Helsinki, Finland
#These authors contributed equally.
This retrospective registry-based cohort study aimed to estimate the incidence and prevalence of genodermatoses in the Swedish population and to analyse associated healthcare usage. Patients diagnosed with genodermatoses were identified from the patient registry of Sahlgrenska University Hospital (Gothenburg, Sweden) between 2016 and 2020. Clinical data from medical records were used to verify diagnoses recorded in the National Patient Registry (NPR). The NPR was then searched for International Classification of Diseases, Tenth Revision (ICD-10) codes Q80–82 and Q84 from 2001 to 2020. The local cohort included 298 patients with 36 unique genodermatosis diagnoses. Verification of these diagnoses in the NPR showed positive predictive values of over 90%. The NPR search yielded 13,318 patients with 73 unique diagnoses, including ichthyoses (n = 3,341; 25%), porokeratosis (n = 2,277; 17%), palmoplantar keratodermas (n = 1,754; 13%), the epidermolysis bullosa group (n = 1011; 7%); Darier disease (n = 770; 6%), Hailey-Hailey disease (n = 477; 4%) and Gorlin syndrome (n = 402; 3%). The incidence and prevalence of each diagnosis were calculated based on the nationwide cohort and are reported. A total of 149,538 outpatient visits were registered, a mean of 4.6 visits per patient. This study provides a valuable resource for the epidemiology of genodermatoses by reporting on the incidence and prevalence of 73 different genodermatoses.
SIGNIFICANCE
Healthcare systems may be unaware of the healthcare requirements and numbers of patients with rare diseases, including genetic diseases. This study provides important insights into the incidence, prevalence, and healthcare utilization associated with genodermatoses in Sweden. The large sample size and comprehensive data collection methods used contribute to a better understanding of the epidemiology of 73 different genodermatoses, including some of the most prevalent, such as ichthyoses and porokeratosis. The positive predictive values of over 90% for the diagnoses verify the accuracy of the findings. This information can be used to improve healthcare planning and resource allocation for patients with genodermatoses.
Key words: epidemiology; genetic skin disease; ichthyosis; incidence; neurofibromatosis; porokeratosis.
Citation: Acta Derm Venereol 2023; 103: adv12404. DOI https://doi.org/10.2340/actadv.v103.12404.
Copyright: © Published by Medical Journals Sweden, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/)
Accepted: Jul 11, 2023; Published: Aug 24, 2023
Corr: Sirkku Peltonen, Department of Dermatology and Venereology, Institute of Clinical Sciences, Sahlgrenska Academy, University of Gothenburg, Gröna stråket 16 Pl 2 SU/Sahlgrenska, SE-41345 Gothenburg, Sweden. E-mail: sirkku.peltonen@gu.se
Competing interests and funding: The authors have no conflicts of interest to declare.
INTRODUCTION
Hereditary skin diseases, also called genodermatoses, are caused by pathogenic variants in various genes expressed in the skin. The current number of genodermatoses is well over 700, but the exact number is not known. In 2009, 560 distinct disorders were defined as genetic skin diseases in the Online Mendelian Inheritance in Man® (OMIM®) database (1, 2). These disorders were associated with 501 unique protein-encoding genes; the rapid development of next generation sequencing (NGS) technology has resulted in yearly new discoveries (3). Although new genodermatoses are continuously being described, only approximately 100 genodermatoses are listed in the International Classification of Diseases, Tenth Revision (ICD-10) (4). The ICD-10 is widely used to register healthcare data, and forms a basis for the allocation of healthcare resources, for disease-based money transfers, and for insurance benefits for patients.
In the ICD-10, the genodermatoses are divided into blocks: congenital ichthyosis; epidermolysis bullosa (EB); other congenital malformations of skin; and other congenital malformations of the integument. Certain syndromes with important skin findings, including neurofibromatosis and Gorlin syndrome, are also classified as phacomatoses or other specified congenital malformation syndromes affecting multiple systems, and there are several other rare diseases scattered in various chapters of the ICD-10, which have a genetic background and skin findings.
According to the European Union’s definition, rare diseases are conditions with an incidence < 1:2,000 (5). Most genodermatoses are rare diseases, but their incidence or prevalence is often unknown. The estimated incidence of genodermatoses varies from 1:250 for ichthyosis vulgaris to 1:500,000 for lamellar ichthyosis (6, 7). On the other hand, the estimates for the same disease may vary considerably, e.g. from 1:30,000 to 100,000 for Darier disease (8).
The genodermatoses exhibit great phenotypical variability, from mild to life-threatening. The most severe genodermatoses, including some subtypes of EB and ichthyosis, have gained more attention in the medical literature than others. For example, a search in PubMed returns 7 times more studies on EB or ichthyosis than on porokeratosis. Most studies on genodermatoses concentrate on a single disease or a group of closely related diseases, and studies on diagnosis, pathogenesis or treatment predominate in number over epidemiological studies. Large registry studies of rare diseases are, nevertheless, important, as they may reveal circumstances not otherwise detectable, e.g. increased mortality and comorbidities (9–11).
Nationwide registries provide an opportunity to form larger study cohorts on rare diseases (12, 13). The national patient registry (NPR), hosted by the Swedish National Board of Health and Welfare, collects information on all inpatient and outpatient visits and uses the Swedish version of ICD-10 diagnosis coding (ICD-10-SE). The diagnoses are automatically transferred from hospitals to the NPR, and positive predictive values (PPVs) of the NPR have previously been shown to be high (85–95%) (14). Diagnosis validation studies on EB and ichthyoses have been carried out recently using the Danish National Patient Registry (15, 16). These studies have analysed the PPVs of first-time diagnoses of ichthyosis and EB made at specialized dermatological departments in the Danish National Patient Registry. The validity of first-time diagnoses of EB ranged from low to average, but for the most common ichthyoses it was over 80%.
The primary aim of this study was to investigate the occurrence of genodermatoses at the Department of Dermatology and Venereology at Sahlgrenska University Hospital (SUH) and to use this data to validate the diagnostic precision in the NPR. The incidence rates of the ICD-10-SE coded genodermatoses, as recorded in the NPR, were studied in the Swedish population. The secondary aim was to benchmark the healthcare usage for this patient group by analysing the number of doctor’s appointments in all hospital departments of dermatology in Sweden.
MATERIALS AND METHODS
Local patient cohort
The study was carried out with the approval of the Swedish Ethical Review Authority (approval number 2020–06202) and SUH. The catchment population of SUH covers the Gothenburg area in Sweden and comprises approximately 750,000 residents. The digital patient registry of SUH was first searched for patients who had had an outpatient visit at the Department of Dermatology and Venereology at SUH and who had received an ICD-10-SE diagnosis corresponding to a genodermatosis (i.e. Q80–Q82, excluding Q82.2 – mastocytosis and Q82.5 – congenital non-neoplastic nevus), a phacomatosis (Q85 for neurofibromatosis and tuberous sclerosis) or Q87.5 to cover Gorlin syndrome. This search covered the time-period from 1 January 2016 to 31 December 2020. The patient records were reviewed for clinical data to validate the diagnosis and to identify any misdiagnoses or clerical errors. The evaluation was based on clinical descriptions, photographs, histopathology, family history and molecular genetic analyses. The numbers of physicians’ appointments were retrieved.
The unique personal identity number, which is assigned to all permanent residents in Sweden, was used to link the local patient cohort to the diagnoses and associated patients registered in the NPR. The PPVs were calculated for all diagnoses represented in the cohort and separately for porokeratosis, EB, ichthyosis, Darier disease and Hailey-Hailey disease by dividing the number of true positives by the number of true positives plus false positives (i.e. true positive / (true positive + false positive)). True positive was defined as SUH positive and NPR positive, while false positive was defined as SUH negative and NPR positive.
Nationwide patient cohort
To collect nationwide data, the NPR for both inpatients and outpatients was searched for ICD-10-SE codes Q80–Q82 (ichthyoses and EB), Q84 (congenital malformations related to skin or skin appendages) and Q87.5 (other congenital malformation syndromes with other skeletal changes) for Gorlin syndrome in the time period from 1 January 2016 to 31 December 2020. The diagnosis codes Q82.2 (mastocystosis) and Q82.5 (congenital non-neoplastic nevus) were excluded for the purposes of this study. The number of patients with each of the ICD-10-SE codes and the number of outpatient visits were retrieved. The incidences of the diseases or closely related disease groups were calculated.
RESULTS
Local patient cohort
A total of 297 patients had been registered with a diagnosis of a hereditary skin disease at the Department of Dermatology and Venereology SUH over the period 2016 to 2020. There were 179 (60%) female and 118 (40%) male individuals, and the total number of different diagnoses was 36. The diagnosis was made for the first time for 226 patients in 2016 to 2020, and the age at diagnosis ranged from 1 day to 94 years. The median age at the first visit was 45 years.
The distribution of diagnoses in the local cohort is shown in Table I and Fig. 1. The largest patient group, 39%, was that of porokeratosis, with 117 patients. The second most common was neurofibromatosis (n = 32; 11%), of which 31 had neurofibromatosis 1, and 1 had neurofibromatosis 2. Ichthyosis was the third largest group (n = 26; 9%), followed by Gorlin syndrome (n = 18; 6%) and Hailey-Hailey disease (n = 15; 5%). Darier disease (n = 13), and EB (n = 12) totalled approximately 4% each. No Q diagnoses or evidence of a genodermatosis was found in 18 of the patient files. Almost two-thirds (21/36) of diagnoses were represented only by 1 or 2 patients, and 20% of all patients had 1 of these rarest diseases. There were several diagnoses with only 1 patient, especially among the ichthyoses and in the group of Other congenital malformations of skin, classified in ICD-10-SE as Q82.
| ICD-10 | Swedish ICD-10-SE | Name of disorder/diagnosis | Patients, n |
| Q80.0–Q80.9 | Q80.0–Q80.9 | Ichthyosis | 26 |
| Q810–Q81.9 | Q81.0–Q81.9 | Epidermolysis bullosa | 12 |
| Q82.8 | Q82.8D | Benign familial pemphigus (Hailey-Hailey disease) | 15 |
| Q82.8E | Keratosis follicularis (Darier’s disease) | 13 | |
| Q82.8F | Inherited keratosis palmaris et plantaris | 10 | |
| Q82.8 | Q828H–Q82.8R and Q82.8W * | 10 | |
| Q828T | Porokeratosis (Mibelli) | 117 | |
| Q82.9 | Q82.9 | Congenital malformation of skin, unspecified | 3 |
| Q85.0 | Q85.0** | Neurofibromatosis | 32 |
| Q85.1 | Tuberous sclerosis | 15 | |
| Q85.8–Q85.9 | Other phakomatoses, not elsewhere classified and Phakomatosis, unspecified | 5 | |
| Q87 | Q87 | Other specified congenital malformation syndromes affecting multiple systems | 15 |
| Q87.5 | Q87.5 | Other congenital malformation syndromes with other skeletal changes, Gorlin syndrome | 18 |
| No diagnosis | 6 | ||
| Total | 297 | ||
| *1–3 patients in each of these diagnostic categories: cutis verticis gyrata, hemangiomatosis (systemic), hidrotic ectodermal dysplasia, keratosis follicularis spinulosa decalvans, pseudoxanthoma elasticum, acrokeratosis verruciformis (Hopf), other congenital malformation of skin. **Contains neurofibromatosis 1 and 2. | |||
| ICD-10: International Classification of Diseases, Tenth Revision. | |||
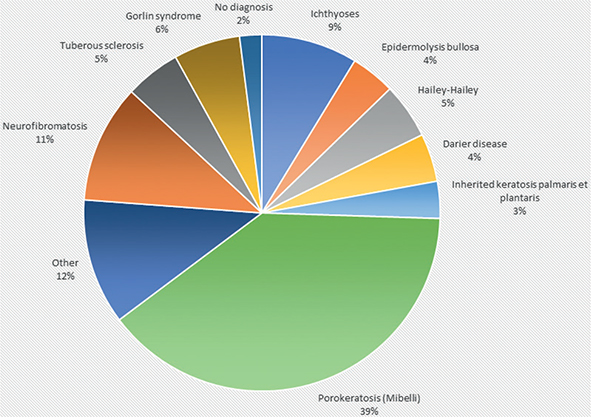
Fig. 1. Distribution of the most common hereditary skin diseases treated at Sahlgrenska University Hospital in 2016 to 2020.
The diagnosis of most patients was based on clinical findings. Of all patients, 16% (n = 49) had undergone skin biopsy for diagnostic purposes during follow-up. Molecular diagnosis was performed for 13% (n = 39) of the patients. Various methods were used: carrier testing for a known familial pathogenic variant (5 patients); Sanger sequencing of single genes; gene panels using NGS; and whole-exome sequencing (2 patients who were siblings). Overall, 31 patients/families received genetic counselling at the Department of Clinical Genetics and Genomics of SUH.
To estimate the use of healthcare services by the patients with genodermatosis, the numbers of outpatient doctors’ appointments during the follow-up period were collected. Overall, patients with genodermatoses made a total of 748 outpatient visits, with a mean of 2.51 visits per patient. Of these, 184 patients (61.5 %) had only 1 registered visit and 79 patients (26.5 %) had 2–4 visits. Sixteen patients ≥ 10 visits, and, collectively, this group made 112 visits, which comprises 15% of the total number of visits.
Of all Q-diagnoses at the Department of Dermatology and Venereology of SUH, 90% were true positives (recorded in the NPR). The PPV for all the diagnoses was 96.1% (95% confidence interval (CI), 92.4–98.2%). It was 90.9% (95% CI, 62.3–98.4%) for Darier disease, 93.1% (95% CI, 86.2–96.8) for porokeratosis, and 100% for ichthyosis, for EB, for Hailey-Hailey disease and for Gorlin syndrome.
Nationwide patient cohort
In the nationwide cohort, 13,318 individuals (Table II) were identified for analysis of incidence and prevalence of hereditary skin diseases in the Swedish population. They had a total of 73 genodermatosis diagnoses in NPR (Table III). The distribution of the most common hereditary skin diseases in Sweden are shown in Fig. 2. The largest patient group was ichthyosis, with a total of 3,341 patients (25% of the total number of patients). The second largest patient group was porokeratosis (n = 2,277; 17%), followed by hereditary keratosis palmaris et plantaris (n = 1,754; 13%), and EB (n = 1011; 7%). There were 770 patients (6% of the total) with Darier disease, 477 with Hailey-Hailey disease (4% of the total), and 402 (3%) with Gorlin syndrome. Since patients could have more than 1 Q diagnosis, the total number of patients is greater than the number of unique patients.
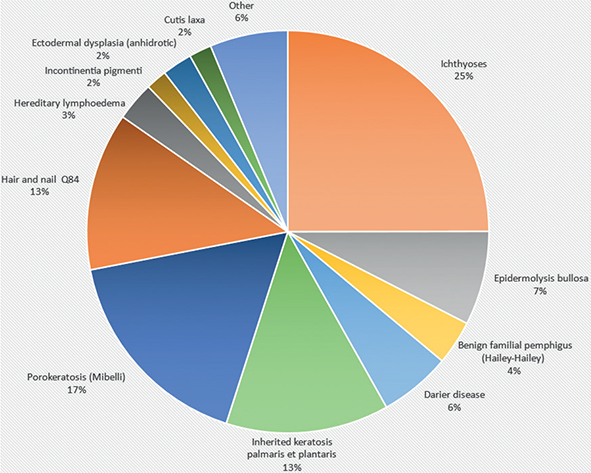
Fig. 2. Distribution of the most common hereditary skin diseases in Sweden in 2001 to 2020.
The most common hereditary skin disease in the nationwide cohort was porokeratosis with a prevalence of 1:4,000, followed by hereditary keratosis palmaris et plantaris at 1:5,000. In the ichthyosis patient group, Q80.9 (congenital ichthyosis, unspecified) was the most common diagnosis, followed by ichthyosis vulgaris (Q80.0), lamellar ichthyosis (Q80.2), and X-linked ichthyosis (Q80.1). Rarer forms of ichthyosis, such as ichthyosis linearis circumflexa and ichthyosis hystrix, were also recorded in ICD-10-SE. In the EB patient group, the most common diagnosis was Q81.9 (EB, unspecified). EB simplex (Q81.0) was the second most common diagnosis, followed by EB dystrophica (Q81.2), and EB letalis (Herlitz syndrome Q81.1).
Overall, a total of 149,538 outpatient visits were registered; a mean of 4.6 visits per patient. The diagnosis with the largest number of visits per patient was Q87.5 (representing Gorlin syndrome) with 10.2 visits per patient. The mean number of outpatient visits and the prevalence and incidence of genodermatoses in the nationwide cohort are shown in Table III.
DISCUSSION
The incidence and prevalence of most genodermatoses are, at best, rough estimates based on data from single centres. For several rare genodermatoses no reliable data exists. The current study reports the incidence and prevalence of 73 hereditary skin diseases in a nationwide cohort, illustrating the relative frequencies.
Two patient cohorts were included in this study. The first was a local cohort of 297 patients from a single dermatological clinic in a university hospital. This cohort also included patients with phacomatoses and Gorlin syndrome. This cohort was used to validate the diagnoses in the NPR and to collect detailed data on, for example, age at diagnosis and diagnostic methods. The second cohort was nationwide and consisted of 13,318 persons who had visited secondary referral centres for hereditary skin disease within public healthcare.
Scrutiny of the patient records of the local cohort and comparisons between the diagnoses of this cohort and the diagnoses in the NPR showed that the PPV for all diagnoses was high (over 90%), suggesting that the diagnoses in the nationwide cohort are reliable.
As in other university hospitals in Sweden, the patients attend the Department of Dermatology in SUH by referral from primary healthcare, regional hospitals, private specialist clinics and other units in the hospital. Thus, the data of the local cohort represents an accurate description of the patients with genodermatoses seen within specialized healthcare, and the results can be generalized to other dermatology referral centres.
The emphasis of dermatology services is on the first visit; the follow-up of most patients takes usually place in the referring unit. This explains why most patients had only 1–4 outpatient visits. However, there is a subgroup of 5.4% (16/297) of the patients who required ≥10 visits. This may be interpreted as a sign of a severe disease, difficulties in finding satisfactory treatments and/or treatment that requires specialist follow-up, e.g. acitretin medication or multiple skin cancers.
This study noted a substantial unmet need for molecular diagnostics, since only 39/297 (13%) patients in the local cohort had a molecular diagnosis. The diagnosis of hereditary skin diseases has traditionally been based on clinical findings supported with histopathology. The availability of NGS-based gene panels has increased the likelihood of getting a molecular diagnosis for hereditary skin diseases in a fraction of the time and at more affordable cost. It seems that a change of thinking among clinicians, wider availability of clinical genetics services, and a redistribution of budget resources are needed to increase the use of molecular diagnostics.
The relative occurrence of various diagnoses in the local cohort yielded somewhat surprising results: the 2 most common diagnoses were porokeratosis and neurofibromatosis. In the current edition of ICD-10 there is only a diagnostic code for porokeratosis (Mibelli), but there are several clinical types of porokeratosis hereditary in an autosomal dominant manner (2, 17, 18). Thus, hereditary porokeratosis would be a more correct term. We have recently shown that porokeratosis is a common genodermatosis (18).
The second largest diagnostic group in the local cohort was neurofibromatosis, with 32 clinically verified diagnoses. The patients had been referred either for diagnosis or for removal of cutaneous neurofibromas with CO2 laser. Phacomatoses, including neurofibromatosis, were not included in the nationwide search because the ICD-10-SE version has only one code, Q85.0, for both neurofibromatosis 1 and neurofibromatosis 2, and previous studies from Finland showed that healthcare registers include many false diagnoses for neurofibromatosis 1 (19).
Ichthyosis and EB are prominent diseases, as judged by the number and impact of publications, which may be due to the presence of severe forms of these diseases. Based on a previously reported estimate of a high (1:250) prevalence of ichthyosis vulgaris (7), we expected ichthyoses to be the most common genodermatosis in the cohort, but this was not the case, since only 24 patients (8% of the total number of patients) had this diagnosis in the local cohort. A possible explanation for this is probably related to the fact that most patients with ichthyosis vulgaris are treated within the primary healthcare or that this genodermatosis is misdiagnosed as atopic dermatitis. The relative number of patients with EB in the whole cohort was lower than expected. It is possible that patients with mild EB are missing, since, without molecular diagnostics, these patients may not have received a correct diagnosis and were not included. The dominant diagnostic subclassification was “unspecified” of both the ichthyoses and EB, which could be due to clinicians not being able to identify the subtypes of EB and ichthyosis appropriately in the first patient examination.
Both Darier disease and Hailey-Hailey disease were more common in the current study than previously estimated (20, 21), with a prevalence of 1:12,000 and 1:19,000, respectively. Both are hereditary in an autosomal dominant manner. Since the conditions become manifest in the third decade of life, they may be less visible than childhood-onset genodermatoses, but because they are completely penetrant, they may reduce the functionality of these patients who then seek medical attention with a low threshold.
Hereditary keratosis palmaris et plantaris was another common genodermatosis in the study, with an estimated prevalence of 1:5,000. With the advent of molecular diagnostics with NGS-based gene panels, hereditary keratosis palmaris et plantaris may be more heterogeneous than previously thought, and a more robust molecular-based classification may emerge. Since there are also very mild cases, it is possible that full coverage of this disease is not possible (22).
Keratosis follicularis decalvans is a rare, X-linked disease with specific ichthyosis and loss of hair (23, 24). The local cohort included 1 patient, but, surprisingly, in the NPR cohort there were many patients. The register data showed, however, that certain hospitals were over-represented in making this diagnosis and approximately half of the patients were women. Thus, we decided not to report the incidence and prevalence of keratosis follicularis decalvans.
In the nationwide data there were 9 genodermatoses that had only been diagnosed in 1 or 2 individuals. This could be interpreted as a sign that some hereditary skin diseases are ultra-rare in the clinical setting. Alternatively, incorrect diagnostics is possible, and this highlights the need for more accurate, molecular-based diagnostics in the field of hereditary skin diseases.
One of the drawbacks of the current study is incomplete ascertainment. Since referrals for mild genodermatosis may be redirected from university clinics to private dermatologists without an assessment in the university clinic, it is possible that the study has not included all patients with genodermatosis. The nationwide cohort was pseudonymized before it was delivered to us, and thus we could not validate the diagnoses from patient journals. Performing epidemiological studies on rare diseases is challenging because the number of patients is low, and most diseases do not have specific diagnosis codes. Since the genodermatoses represent a variety of diseases with different expressions and occurrences, there may be diagnostic delays and uncertainties in the recording of the diagnosis. Correspondingly, the diagnostic accuracy in clinical work may not always be sufficient for making proper diagnoses of rare diseases.
Patients with rare diseases may be invisible in clinical centres that do not have a specific interest in rare diseases. It is likely that the number of patients and their service usage or needs are not recognized even if they have chronic symptoms and decreased functionality. This study provides a unique resource for the epidemiology of genodermatoses, since it is mandatory to report diagnoses to the NPR from all public hospitals in Sweden, enabling the extraction of high-quality epidemiological data. The findings of this study will be of use for benchmarking the resources required for this patient group, including doctors and nurses, as well as genetic counselling.
ACKNOWLEDGEMENTS
The study was funded by grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement, University of Gothenburg, and by the HudFonden.
The Swedish Ethical Review Authority approved the study. Research permissions were obtained from the Sahlgrenska University Hospital, the Swedish National Board of Health and Welfare and Statistics Sweden. The study followed the principles of the Declaration of Helsinki. The study was register-based and retrospective and therefore exempt from obtaining informed consent from the participants. All analyses were carried out using pseudonymized data.
Chat Generative Pre-Trained Transformer (ChatGPT) Version 3.5 was used to improve the text in the Significance paragraph.
REFERENCES
- Feramisco JD, Sadreyev RI, Murray ML, Grishin NV, Tsao H. Phenotypic and genotypic analyses of genetic skin disease through the Online Mendelian Inheritance in Man (OMIM) database. J Invest Dermatol 2009; 129: 2628–2636.
- Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins Univer sity (Baltimore, MD), 2023. Available from: https://omim.org.
- Chiu FP, Doolan BJ, McGrath JA, Onoufriadis A. A decade of next-generation sequencing in genodermatoses: the impact on gene discovery and clinical diagnostics. Br J Dermatol 2021; 184: 606–616.
- International Statistical Classification of Diseases and Related Health Problems 10th Revision. World Health Organization, 2019. Available from: https://icd.who.int/browse10/2019/en.
- European Commission; an official website of the European Union. Rare Diseases. Available from: https://ec.europa.eu/health/non-communicable-diseases/steering-group/rare-diseases_en.
- Krug M, Oji V, Traupe H, Berneburg M. Ichthyoses – part 2: congenital ichthyoses. J Dtsch Dermatol Ges 2009; 7: 577–588.
- Wells RS, Kerr CB. Clinical features of autosomal dominant and sex-linked ichthyosis in an English population. Br Med J 1966; 1: 947–950.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol 2003; 4: 97–105.
- Curman P, Bern J, Sand L, Cederlöf M, Bachar-Wikström E, Wikström JD. Patients with Darier Disease exhibit cognitive impairment while patients with Hailey-Hailey disease do not: an experimental, matched case-control study. Acta Derm Venereol 2021; 101: adv00476.
- Kallionpää RA, Valtanen M, Auranen K, Uusitalo E, Rinne JO, Peltonen S, et al. Increased risk for dementia in neurofibromatosis type 1. Genet Med 2021; 23: 2219–2222.
- Ahanian T, Curman P, Leong IUS, Brismar K, Bachar-Wikstrom E, Cederlöf M, et al. Metabolic phenotype in Darier disease: a cross-sectional clinical study. Diabetol Metab Syndr 2020; 12: 12.
- Ludvigsson JF, Otterblad-Olausson P, Pettersson BU, Ekbom A. The Swedish personal identity number: possibilities and pitfalls in healthcare and medical research. Eur J Epidemiol 2009; 24: 659–667.
- Bachar-Wikström E, Wikström JD. Darier disease – a multi-organ condition? Acta Derm Venereol 2021; 101: adv00430.
- Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim JL, Reuterwall C, et al. External review and validation of the Swedish national inpatient register. BMC Public Health 2011; 11: 450.
- Kristensen MH, Schmidt SAJ, Kibsgaard L, Hove H, Sommerlund M, Koppelhus U. Validity of first-time diagnoses of inherited ichthyosis in the Danish National Patient Registry and the Danish Pathology Registry. Clin Epidemiol 2020; 12: 651–657.
- Kristensen MH, Schmidt SAJ, Kibsgaard L, Mogensen M, Sommerlund M, Koppelhus U. Validity of first-time diagnoses of congenital epidermolysis bullosa in the Danish National Patient Registry and the Danish Pathology Registry. Clin Epidemiol 2019; 11: 115–124.
- Kubo A, Sasaki T, Suzuki H, Shiohama A, Aoki S, Sato S, et al. Clonal expansion of second-hit cells with somatic recombinations or C>T transitions form porokeratosis in MVD or MVK mutant heterozygotes. J Invest Dermatol 2019; 139: 2458–66.e9.
- Inci R, Zagoras T, Kantere D, Holmström P, Gillstedt M, Polesie S, et al. Porokeratosis is one of the most common genodermatoses and is associated with an increased risk of keratinocyte cancer and melanoma. J Eur Acad Dermatol Venereol 2023; 37: 420–427.
- Uusitalo E, Leppävirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol 2015; 135: 904–906.
- Tavadia S, Mortimer E, Munro CS. Genetic epidemiology of Darier’s disease: a population study in the west of Scotland. Br J Dermatol 2002; 146: 107–109.
- Ben Lagha I, Ashack K, Khachemoune A. Hailey-Hailey disease: an update review with a focus on treatment data. Am J Clin Dermatol 2020; 21: 49–68.
- Harjama L, Karvonen V, Kettunen K, Elomaa O, Einarsdottir E, Heikkilä H, et al. Hereditary palmoplantar keratoderma – phenotypes and mutations in 64 patients. J Eur Acad Dermatol Venereol 2021; 35: 1874–1880.
- Verma R, Bhatnagar A, Vasudevan B, Kumar S. Keratosis follicularis spinulosa decalvans. Indian J Dermatol Venereol Leprol 2016; 82: 214–216.
- Cantu JM, Hernandez A, Larracilla J, Trejo A, Macotela-Ruiz E. A new X-linked recessive disorder with dwarfism, cerebral atrophy, and generalized keratosis follicularis. J Pediatr 1974; 84: 564–567.