SHORT COMMUNICATION
Familial Cutaneous Angiosarcoma of the Head
Masakazu KAKURAI, Shusaku ITO*, Akemi MAEDA, Daiki OGAWA and Rie HONDA
Division of Dermatology, Hitachi General Hospital, 2-1-1 Jonan, Hitachi, Ibaraki 317-0077, Japan. *E-mail: shusaku.ito.ku@hitachi.com
Citation: Acta Derm Venereol 2023; 103: adv15314. DOI: https://doi.org/10.2340/actadv.v103.15314.
Copyright: © Published by Medical Journals Sweden, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/)
Accepted: Oct 3, 2023; Published: Nov 21, 2023
INTRODUCTION
Angiosarcoma (AS) is a highly aggressive malignant tumour that accounts for approximately 2% of soft tissue sarcomas (1–3). The familial occurrence of AS is extremely rare. To the best of our knowledge, there are 4 cases of familial cardiac AS in the literature (4–7), but none of familial cutaneous AS. We report here 2 cases of cutaneous AS of the head that developed in a father and then in his son within 1 year.
CASE REPORT
Case 1. A 79-year-old Japanese man with a lesion on the scalp for 1 month that started to bleed was referred to Hitachi General Hospital, Ibaraki, Japan. He had a medical history of colon cancer that was treated with resection at the age of 69 years. A physical examination showed a mass measuring 45×25 mm with blood crusts and surrounded by irregularly shaped purpura on the right crown region of the scalp (Fig. 1a). Skin biopsy of the mass revealed atypical endothelial cells lining irregular vascular spaces and haemorrhages in the dermis (Fig. 1b). Tumour cells were positive for CD31 and D2-40 and negative for CD34 and human herpesvirus-8 (HHV-8) (Fig. 1c–f). These findings corroborated the diagnosis of cutaneous AS. Metastasis was not observed on computed tomography. Radiotherapy was delivered at 70 Gy in 35 fractions over 7 weeks and paclitaxel at a dose of 60 mg/m2 was administered on days 1, 8 and 15 in 4-week cycles for 11 months. Due to a decline in the activities of daily living, paclitaxel was discontinued. Metastasis was not detected 1 year after the initiation of chemoradiation therapy. Five months after the cessation of paclitaxel, the patient died of COVID-19 infection.
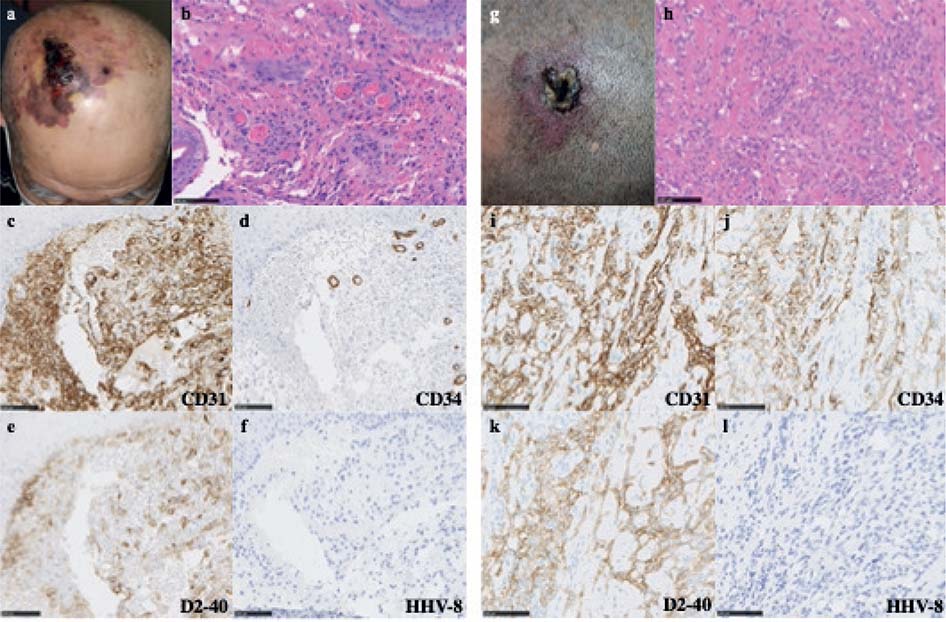
Fig. 1. Clinical and histological findings. (a) Mass and purpura on the right crown region of the scalp. (b) Haematoxylin and eosin (H&E) staining of a biopsy specimen of the mass revealed atypical spindle-shaped cells forming irregular vessels in the dermis. (c–f) Tumour cells were positive for CD31 and D2-40 and negative for CD34 and human herpesvirus-8 (HHV-8). (g) Plaque on the left frontal region of the scalp. (h) H&E staining of a biopsy specimen showed atypical cells forming dilated irregular vascular structures in the dermis. (i–l) Tumour cells were positive for CD31, CD34 and D2-40 and negative for HHV-8. Bars: 100 μm.
Case 2. A 50-year-old Japanese man, the son of case 1, with a history of hypertension had a purple spot on the scalp for 4 months. He was referred to our hospital within 1 year of his father’s visit. None of his other family members, including his mother and younger brother, had AS. A physical examination showed an irregularly shaped red-purple plaque measuring 42×32 mm with blood crusts on the left frontal region of the scalp (Fig. 1g). A biopsy specimen from purpura on the left frontal region of the scalp showed atypical cells forming dilated irregular vascular structures in the dermis (Fig. 1h). Tumour cells were positive for CD31, CD34 and D2-40 and negative for HHV-8 (Fig. 1i–l). Based on these findings, the patient was diagnosed with cutaneous AS. Metastasis was not observed on positron emission tomography-computed tomography. Since the lesion required extensive resection to achieve a sufficient margin, chemoradiation therapy was selected after obtaining informed consent. Radiotherapy was delivered at 70 Gy in 35 fractions over 7 weeks and paclitaxel at a dose of 60 mg/m2 was administered on days 1, 8 and 15 in 4-week cycles for 16 months. Metastasis was not detected 1 year after the initiation of chemoradiation therapy.
DISCUSSION
AS is a rare and highly aggressive malignant tumour that originates from lymphatic or vascular endothelial cells (1–3). The most commonly affected sites are the skin (49.6%), including head and neck areas and the breast, followed by the parenchyma of the breast (14.4%), soft tissue (11.2%) and the heart (6.7%) (2). To the best of our knowledge, familial cutaneous AS has not been reported in the English or Japanese literature. To date, 4 cases of familial cardiac AS have been reported with an earlier onset than that of sporadic AS (4–7). Two of the 4 cases had the clinical characteristics of Li-Fraumeni-like (LFL) syndrome with a protection of telomeres 1 gene mutation, which is responsible for the development of cardiac AS and other tumour types, such as familial chronic lymphocytic leukaemia, glioma and malignant melanoma (6, 7). Since genetic testing was not performed on the current cases and the father developed the lesion at the age of 79 years old, the diagnosis of LFL syndrome was not confirmed. However, due to the earlier onset of AS in the son than cutaneous AS, which is reportedly a mean age of 73 years (3), we cannot rule out a potential relationship between familial cutaneous AS and genetic factors.
Several risk factors have been reported for AS, including chronic lymphoedema, radiotherapy, toxic chemical exposure and genetic disorders (1–3). Moreover, since HHV-8 has a tropism for endothelial cells, its role in the pathogenesis of AS has been examined (8). However, a previous study concluded that HHV-8 was not involved in the pathogenesis of AS because none of the 14 AS patients tested were positive for HHV-8 (8). The current cases were also negative for HHV-8, indicating no relationship with AS.
In conclusion, we report here the first case of familial cutaneous AS. Although the relationship between familial cutaneous AS and genetic factors remains unclear, familial cutaneous AS is highly notable in the current cases.
REFERENCES
- Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol 2010; 11: 983–991.
- Lahat G, Dhuka AR, Hallevi H, Xiao L, Zou C, Smith KD, et al. Angiosarcoma: clinical and molecular insights. Ann Surg 2010; 251: 1098-1106.
- Albores-Saavedra J, Schwartz AM, Henson DE, Kostun L, Hart A, Angeles-Albores D, et al. Cutaneous angiosarcoma. Analysis of 434 cases from the Surveillance, Epidemiology, and End Results Program, 1973–2007. Ann Diagn Pathol 2011; 15: 93–97.
- Casha AR, Davidson LA, Roberts P, Nair RU. Familial angiosarcoma of the heart. J Thorac Cardiovasc Surg 2002; 124: 392–394.
- Keeling IM, Ploner F, Rigler B. Familial cardiac angiosarcoma. Ann Thorac Surg 2006; 82: 1576.
- Calvete O, Martinez P, Garcia-Pavia P, Benitez-Buelga C, Paumard-Hernández B, Fernandez V, et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-Fraumeni-like families. Nat Commun 2015; 6: 8383.
- Calvete O, Garcia-Pavia P, Domínguez F, Bougeard G, Kunze K, Braeuninger A, et al. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur J Hum Genet 2017; 25: 1278–1281.
- Avancini J, Sanches JA, Cherubim AP, Pazzini R, Oliveira CM, Sumita LM, et al. Angiosarcoma in HIV-negative patients is not associated with HHV-8. An Bras Dermatol 2016; 91: 738–741.