Table I. Clinical and biological findings in patients with ichthyosis, sclerosing cholangitis, acquired alopecia
1Department of Dermatology, Reference Center for Genodermatoses and Rare Skin Diseases (MAGEC-Necker), 2Department of Molecular Genetics, 5Department of Pediatric Gastroenterology-Hepatology-Nutrition, Necker–Enfants Malades University Hospital, Assistance Publique – Hôpitaux de Paris-Centre (AP-HP), 3UMR1163 Intestinal Immunity, Imagine Institut, 4Paris University, Paris, 6Department of Dermatology, Reference Center for Genodermatoses and Rare Skin Diseases (MAGEC-Saint Louis), Hôpital Saint-Louis, Assistance Publique – Hôpitaux de Paris (AP-HP), and 7Institut Imagine. E-mail: smail.hadj@inserm.fr
Accepted May 11, 2020; Epub ahead of print May 19, 2020
Acta Derm Venereol 2020; 100: adv00173
Neonatal ichthyosis-sclerosing cholangitis syndrome (NISCH syndrome, MIM#607626) is a rare autosomal recessive syndromic ichthyosis characterized by scalp hypotrichosis, scarring alopecia, ichthyosis, and sclerosing cholangitis. It is caused by bi-allelic and loss of function mutations in the CLDN1 gene, localized on chromosome 3q27–q28, encoding for claudin-1 (1, 2). Claudin-1 is a 211-amino-acid-long integral membrane protein of tight junction strands, which in humans, is notably expressed in the liver and skin. Tight junctions represent one mode of cell-to-cell adhesion in epithelial or endothelial cell sheets, forming continuous seals around cells and serving as a physical barrier to prevent solutes and water from passing freely through the paracellular space. The syndrome was first described as NISCH syndrome and is now called ichthyosis, leukocyte vacuoles, alopecia, and sclerosing cholangitis (ILVASC).
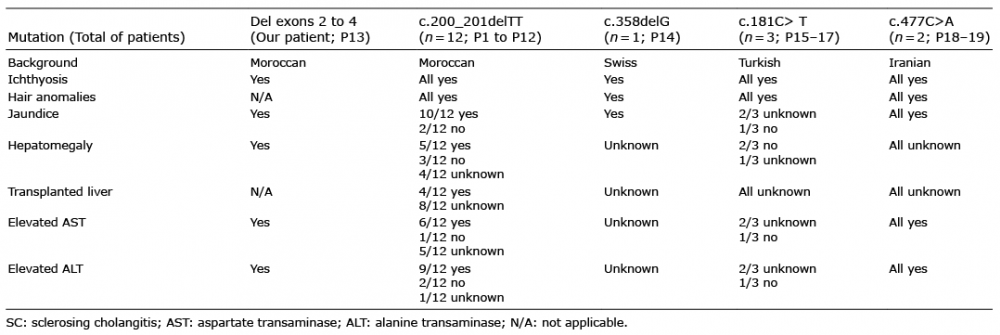
We report here a female neonate presenting with a novel CLDN1 homozygous deletion of exons 2 to 4. She was born of consanguineous parents, with a combination of congenital ichthyosis with fine white scales on the whole body, including; scalp, hypotrichosis, jaundice, moderate hepatomegaly and elevated liver enzymes: aspartate transaminase (AST) 81 IU/l (n < 60), alanine transaminase (ALT) 130 IU/l (n < 40), alkaline phosphatase (ALP) 578 IU/l (n < 380) and conjugated bilirubin 13 micromole/l (n < 5). Gamma-glutamyltransferase (GGT) level was normal. Her parents were heterozygous for the CLDN1 deletion. While reviewing the literature, 19 cases were found, including the current case. It was noted that the most common features associated with CLDN1 mutations are divided mainly into clinical and biological findings. Ichthyosis and hair anomalies; hypotrichosis and/or alopecia were found in all 19 patients, jaundice in 16 patients (84%), oligodontia in 8 (42%), hypodontia in 4 (21%), hepatomegaly in 6 (31%), leucocyte vacuoles in 4 (21%), and a collodion membrane in 2 (10%). All patients had elevated liver enzymes and hyperbilirubinaemia. Cholestasis varied from a transient remitting form to more severe fibrogenic conditions (1–4). Geographically, the 19 patients were from Morocco (13; 68%), Turkey (3; 15%), Iran (2; 10%), and Switzerland (1; 5%) (Table I). To date, 5 different homozygous mutations in 19 patients belonging to 11 families have been reported. Sequence variations were checked and actualized using Alamut® Visual 2.15 software (Interactive Biosoftware, SOPHIA GENETICS company, Rouen, France). Among them 12 patients from Morocco carry the same CLDN1 deletion of 2 base pairs in exon 1 [c.200_201delTT; p.(Phe67*)] suggesting a founder effect for this mutation in Morocco. A second homozygous deletion expressing only exon 1 is reported here, for the first time, in our patient from Morocco. A third homozygous deletion was reported in a Swiss female patient [c.358delG in exon 2; p.(Val120Serfs*15)] (3). The 2 last nonsense mutations were reported in 4 patients from 2 unrelated Turkish families [c.181C>T, p.(Gln61*) in exon 1] and 1 Iranian family [c.477C>A, p.(Tyr159*) in exon 4], respectively (5–7). All these mutations lead to a truncated CLDN1 protein with loss of its transmembrane domains and functions (Fig. 1). DCDC2 mutations were identified in 4 affected children with neonatal sclerosing cholangitis without ichthyosis (8).
Table I. Clinical and biological findings in patients with ichthyosis, sclerosing cholangitis, acquired alopecia

Fig. 1. CLDN1 protein structure and mutations localization. AA: amino acid; c.: codon; p.: protein; P: patient.
By observing the mutations of the Moroccan and the Turkish groups we could assume the existence of 2 founder effects in Morocco and, probably, in Turkey, respectively. Further phenotype comparison showed a positive consanguinity, presence of ichthyosis, presence of hypo-trichosis and/or alopecia in these 2 groups. However, 6 members of the Moroccan group had hepatomegaly, 4 of whom of which had dental anomalies. While neither hepatomegaly nor dental anomalies were identified in the Turkish group. Both groups displayed variable degrees of jaundice in most of their members. The Swiss and Iranian groups shared the same phenotypic characteristics i.e. ichthyosis, hair anomalies and jaundice in all their members, although their mutations happened on different exons, as described earlier (Table I). In all patients, the common phenotype associates ichthyosis, sclerosing cholangitis and acquired alopecia which is probably a consequence of cutaneous inflammation. Thus, rather than ILVASC or NISCH, we suggest simplifying the acronym to ISCAA for ichthyosis, sclerosing cholangitis, and acquired alopecia.
We acknowledge the hard work of Dr. Ahmed S. ZUGAIL, Teaching Assistant, Urology Department, Faculty of Medicine in Rabigh, Jeddah, Saudi Arabia, for his help in the English language editing of this article.