Fig. 1. PRISMA flow diagram.
1School of Medicine, University of Notre Dame, 2St George Hospital, Department of Dermatology, Sydney, Australia, 3Antalya Research and Training Hospital, Antalya, Turkey and 4Faculty of Medicine, University of New South Wales, Sydney, Australia
Bullous pemphigoid is an autoimmune subepithelial disease characterised by pruritus followed by urticarial plaques and finally bullae on the skin and mucosa. Drug-associated bullous pemphigoid (DABP) is a term used to describe instances of bullous pemphigoid demonstrating clinical, histological, or immunopathological features identical or similar to those of the idiopathic form of bullous pemphigoid, associated with the systemic ingestion, or topical application of particular drugs. In this study, we conducted a comprehensive search of the literature according to PRISMA guidelines and a total of 170 publications were included in the final qualitative analysis. In conclusion, 89 drugs were implicated in DABP. The strongest evidence for DABP is seen with gliptins, PD-1/PD-L1 inhibitors, loop diu-retics, penicillin and derivatives. An appreciation of the medications associated with bullous pemphigoid enables clinicians to identify potential cases of DABP earlier and cease the offending medication.
Key words: bullous pemphigoid; drug-associated; drug-associated bullous pemphigoid.
Accepted Mar 12, 2020; E-pub ahead of print Mar 16, 2020
Acta Derm Venereol 2020; 100: adv00224.
Corr: Prof Dédée F. Murrell. Department of Dermatology, St George Hospital, Sydney, NSW 2217, Australia. E-mail: d.murrell@unsw.edu.au
Bullous pemphigoid (BP) is an autoimmune subepi-thelial disease characterised by the generation of pruritus followed by urticarial plaques and finally bullae on the skin and mucosa. BP arises when autoantibodies are generated against two hemidesmosomal proteins, BP230 and BP180. This leads to the activation of the complement cascade, inflammatory cell migration, and formation of subepithelial bullae (1). Drug-associated BP (DABP) is a term used to describe instances of BP demonstrating clinical, histological, or immunopathological features identical or similar to those of the idiopathic form of the disease. DABP may be associated with the systemic ingestion or topical application of particular drugs (2). An appreciation for the underlying genetic susceptibility of the individual to the particular drugs has been postulated, as only a select few experience the clinical disease when exposed, hence suggesting a multifactorial origin of the disease.
In the ensuing review, we will concisely describe the epidemiology, genetic implications, aetiology, pathophysiology of the drug reaction, followed by a comprehensive discussion of the drugs implicated in DABP.
A comprehensive search of the literature was performed according to the PRISMA guidelines using the PubMed, MEDLINE, EMBASE and Cochrane Library, Scopus, ProQuest, and Web of Science databases (3). Search criterion used to facilitate this were: [“Pemphigoid, bullous pemphigoid, drug-induced bullous pemphigoid, drug-induced pemphigoid, bullous/chemically induced pemphigoid*” MeSH]. The search was limited to those published before January 13, 2019. Firstly, 1,191 articles have been found and then further publications were identified through manual evaluation of the references included in the retrieved publications. After removing duplicates, irrelevant articles were excluded. Further on, non-original review, commentaries and publications without full text were excluded. There were 52 articles in languages other than English, full texts were available for 15, which were translated and incorporated into the analysis. Finally, 170 publications were included in this qualitative synthesis (Fig. 1).
Fig. 1. PRISMA flow diagram.
In our literature review, we identified 250 case reports of DABP. These cases implicated 89 individual drugs from 9 diverse classes (Table SI). Each case is incorporated into a detailed spreadsheet revealing the characteristics of the implicated drug (Table SII). Based on the temporal relationship with administration and withdrawal, recurrence with re-challenge, and the diagnostic certainty we were able to gauge the strength of association with the drug.
Epidemiology
BP is the most common autoimmune blistering disorder with a rising incidence of between 4 to 22 new cases per million individuals per year in Europe (4, 5). Observational studies have reported between a 1.9–4.3-fold increase in the number of the cases in the past two decades (4, 6, 7). BP most commonly occurs after the sixth to seventh decade. Younger people are rarely affected and this is often more severe than in the elderly (8). A vast majority of the BP cases occur without an identifiable cause, although recognition of the underlying cause may be fostered through a meticulous history, with an emphasis on inducing factors, such as co-existing conditions, and medications. A distinguishable precipitant is identified in < 15% of patients (9).
Genetic implications in drug-associated bullous pemphigoid
An appreciation of the underlying genetic susceptibility to BP has gradually been realised through an enhanced understanding of the pathogenesis of the disease and novel experimental techniques. An association of BP with certain Major Histocompatibility Complex (MHC) class II alleles has been demonstrated by several investigators. In particular, a significant association with the human leukocyte antigen-DQβ1*0301 allele has been established in the populations of European descent (10, 11). Further studies have revealed that while these findings are applicable to populations of European descent, alternate alleles may be implicated in other populations (12–14). In the Japanese population a significant association with HLA-DRβ1*04, HLA-DRβ1*1101 and HLA-BDB1*0302 alleles has been established (13). The suggested mechanism of these HLA alleles contributing to the susceptibility to BP involves facilitating antigen presentation of basement membrane zone (BMZ) antigens to T cells. These HLA alleles are associated with recognition of conserved epitopes of the antigens of the BMZ in patients with BP contributing to the initiation of autoimmunity (10, 15–17). Studies have also demonstrated that BP180-specific Th1 and Th2 cells are restricted to the HLA-DQB1*0301 allele with the Th1 phenotype present even in individuals without clinical evidence of the disease (18). Whether the predisposing alleles are identical in idiopathic and DABP remains uncertain. However, some drug-associated adverse reactions have demonstrated a relationship with some HLA alleles (19, 20).
Aetiology and pathophysiology of the drug reaction
In general terms, the pathogenesis of the drug-reaction in DABP is beginning to be unravelled, however, the specific causal relationships remain to be elucidated. Drugs are considered to act as triggers in those with an underlying genetic predisposition, leading to either augmentation of the immune response or an alteration of the antigenic properties of the epidermal BMZ (21). Drugs may alter these antigenic properties by binding to molecules in the lamina lucida of the BMZ, thereby acting as neoantigens, and inducing the formation anti-BMZ antibodies (22, 23). Alternatively, they may structurally modify molecules and uncover previously hidden epitopes, hence stimulating the immune response (24, 25). Indeed, Patsatsi et al. (26) revealed an elevated titre of anti-BP180NC16A auto-antibodies in a group of patients receiving systemic medications prior to the development of the disease, when compared to the those receiving no medications, supporting the theory of drug-associated epitope spreading. A ‘two-step’ theory has been hypothesised proposing that two separate drugs may induce the disease with evidence observed in BP, and other related bullous dermatoses (27, 28).
These drugs can be categorised according to the functional groups inherent in their chemical structure, as either thiol-drugs, phenol-drugs, and non-thiol non-phenol drugs.
Thiol-based medications. The vast majority of thiol drugs are known to contain, or release sulfhydryl groups either within the precursors, or the catabolised metabolite (29) (Table I). Thiol-drugs are capable of structurally modifying the molecule to either act as haptens, or uncover epitopes resulting in the formation of anti-BMZ antibodies. Alternatively, through an independent non-immune mechanism, the metabolism of these thiol-drugs is cap-able of disrupting the integrity of the dermo-epidermal junction in the BMZ attained by interaction with the sulfhydryl groups in desmosomes (21, 30). Particular thiol-drugs, for example penicillamine, are able to decrease the activity of regulatory T cells (Treg) enabling the hyperproduction of autoantibodies towards the antigens of the BMZ (2, 31). These mechanisms synergistically promote the initiation, and progression of the disease.

Table I. Medications implicated in drug-associated bullous pemphigoid
Phenol-based medications. Phenol-drugs are those recognised to incorporate a phenyl group bonded to a hydroxy group in their molecular structure. The phenol-drugs associated with DABP include a collection of the cephalosporins, and aspirin (2, 31). These drugs are implicated in other autoimmune blistering diseases, namely pemphigus vulgaris, in which they induce acantholysis, a feature not normally present in the classic pathological description of BP (32). A similar mechanism acting to disrupt the integrity of the BMZ may reveal hidden epitopes enabling autoantibody production. Correspondingly, a causal association with aspirin has been proposed where aspirin may serve as a hapten altering the antigenicity of the lamina lucida or by attaching to cellular target sites leading to formation of autoantibodies (31).
Non-thiol non-phenol medications. An expanding assortment of non-thiol non-phenol drugs are recognised as capable of inducing BP (Table I).
Clinical manifestations of the drug reaction
As opposed to the idiopathic form, the clinical manifestations of DABP are heterogenous, which often delays recognition of the underlying diagnosis (Table II). Patients are often considerably younger than those affected by the idiopathic form. Histological features of DABP encompass a few different findings seen often than idiopathic BP (33). No specific antigens for DABP have been identified, suggesting that the antigens are, at least in part, the same of the ones detected in the idiopathic BP (IBP) (25, 34). Immunofluorescence reveals a similar profile to IBP.
The natural history of DABP is relatively uncertain, although two divergent courses have been identified. These include an acute, self-limited form characterised by definitive resolution following the withdrawal of the supposed drug (21). This form is reminiscent of a true-drug reaction. In contrast, a chronic form may be observed demonstrating all of the characteristic features of the idiopathic-form of BP, which persists even after the removal of the offending agent and may require prolonged therapeutic intervention to establish control (21).
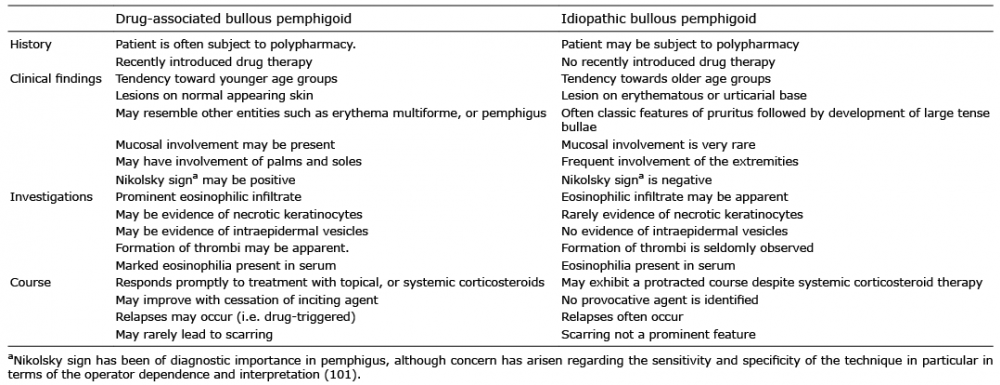
Table II. Observed differences between drug-associated bullous pemphigoid and idiopathic bullous pemphigoid
Drugs implicated in drug-associated bullous pemphigoid
DABP was originally considered to be a distinct disease entity when Bean et al. (22) discovered a bullous eruption following the administration of salicylazosulfapyridine to an 11-year old boy in 1970. Since this initial case 89 drugs have been believed to be associated with inducing BP (Table I and Table SI). While these case reports often describe tentative associations with the administration of the medications based on the temporal relationship, or analogy with previously reported cases, a causal relationship often remains elusive. As such, clinical judgement has regularly been used to justify the diagnosis of drug-associated reactions based on the history, clinical examination, histological and immunopathological findings.
These clinical judgements are subjective to several potentially confounding factors. In most of the cases reported the patients have been exposed to multiple medications simultaneously, thus obscuring the ability to accurately discern the medication responsible (35, 36). Another factor that has come to be appreciated is the influence of over-the-counter medications, and complementary and alternative medicines, which may be withheld either consciously or unconsciously (37, 38). In order to gain more definitive evidence of a causal relationship a rechallenge may be proposed though ethical concerns arise, and therefore many patients who have been rechallenged has been done so though inadvertent exposure to the offending medication rather than a deliberate intervention (38).
Anti-inflammatory drugs. Non-steroidal anti-inflammatory drugs (NSAIDs) were one of the first recognised to be associated with the development of BP (22). There are now 13 cases described in the literature implicating 8 different NSAIDs, which differ in their structural composition and proposed mechanism of inducing BP. Celecoxib contains a sulfonamide moiety, theorised as a hapten through covalent linkages between macromolecules and reactive sulfonamide intermediaries (39). Aspirin is also proposed to act as an autoimmune hapten, notably whilst devoid of a sulfhydryl group, altering the antigenicity of the lamina lucida or to attach to the cellular target site leading to formation of autoantibodies (31). While these case reports demonstrate an association and have proposed mechanisms to explain this, the UK case-control study demonstrated no significant difference in the number of patients taking aspirin in those developing BP (40).
Sulfasalazine contains a sulfhydryl postulated to be responsible for induction of an autoimmune reaction by acting as a hapten to basement membrane entities, and may exhibit a degree of cross-reactivity with dapsone often used for treatment (22).
Rechallenge has been performed with aspirin, diclofenac, phenacetin and sulfasalazine with recurrence further supporting an association (25, 31).
Diuretics. Several classes of diuretics have been implicated in 22 case reports of DABP including the loop diuretics, thiazide diuretics, and aldosterone antagonists (41–44). Loop diuretics, in particular furosemide, have become entrenched in the literature as an inducing agent, even being used effectively as a positive control in case-control studies investigating other agents (45). Loop diuretics are thiol-based drugs enabling the aforementioned immune and non-immune mediated pathogenetic mechanisms to induce BP (2, 21). Similarly, the thiazide diuretics and aldosterone antagonists are also sulfur-containing diuretics resulting in similar pathogenic mechanisms to be proposed (46).
In several case-control studies, a strong association was proposed with aldosterone antagonists (35, 36). Multivariate analysis in these studies demonstrated that the chronic use of spironolactone is significantly associated with BP (OR 2.3–3.1). Nonetheless, in a retrospective case-control study, aldosterone antagonists were not considered to be associated with BP when adjusting for age, sex and co-morbidities (40). Lloyd-Lawery et al. (40) demonstrated in the same study that loop diuretics were associated with BP (adjusted OR 3.8). This finding was also demonstrated for furosemide in the French pharmacovigilance database (reporting OR 3.3). However, this database consisted of spontaneous reporting. These conclusions were in contrast with the earlier studies by Bastuji-Garin et al. that had demonstrated no association with loop diuretics (35, 36). No association with the thiazide diuretics has been demonstrated in these studies. Rechallenge has been performed with furosemide and spironolactone with a similar eruption observed (47, 48).
Cardiovascular (antihypertensives, antiarrhythmics, anticoagulants and statins). Antihypertensives have been implicated as a potential contributing factor in numerous cases of BP, including angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor II antagonists, and calcium channel blockers (CCBs). ACE inhibitors containing sulfhydryl groups, namely captopril, are suspected based on an isolated case report bolstered by a plausible hypothesis of the underlying pathogenesis with a similar molecule, D-penicillamine (49). Enalapril, a non-thiol ACE inhibitor, was recognised as capable of inducing acantholysis in vitro, distinct from that of the hypothesised role of captopril as a hapten, in which acantholysis, may expose components of the BMZ (50). An additional collective mechanism is the potentiation of the kinin pathway through the inactivation of ACE may lead to a proinflammatory state (27). Nonetheless, their hapten-like properties remain more accredited (26).
Both of the CCBs implicated are classed as dihydropyridines, which are primarily selective for the L-type calcium channel. Isolated cases have also occurred with angiotensin II antagonists exposure although several confounding factors obscured their findings results in an ability to exclude an IDP coinciding with administration (51). These findings have not been supported by literature with no significant differences observed between those exposure to antihypertensive, and those who remain unexposed (36, 40). Antiarrhythmic agents have also been described in a limited number of case reports. Those reported include beta-blockers, and unsubstantiated reports of amiodarone (36, 52). These beta-blockers were reported in isolated case reports, but no clear association has been demonstrated in control studies. Rechallenge has been performed with enalapril with prompt recurrence of the lesions observed (34).
Antibiotics. Antibiotics are a relatively rare cause of DABP. Bastuji-Garin et al. originally determined through two French, prospective, case-control studies that no association exists between the use of antibiotics and BP (35, 36). However. this has since been challenged by a UK case-control study which demonstrated that patients with BP used antibiotics significantly more often as compared to control (40). Diverse classes of antibiotics have continued to be reported including the penicillins, cephalosporines, quinolones, nitroimidazoles, actinomycin, and annamycin (Table I). When metabolised penicillins expose a sulfhydryl group that has proposed to be involved in the pathogenesis of the drug reaction (21, 30).
Quinolones are described in 3 case reports of DABP. Levofloxacin does not contain a sulfhydryl group, yet levofloxacin is hypothesised to act as a hapten with evidence of antibodies toward BP180 detected in the affected patients sera (53). An equivalent hypothesis for the pathogenesis has been described for ciprofloxacin (54). Metronidazole, a non-phenol non-thiol antibiotic, has only rarely been associated with bullous eruptions (55)
Antifungal agents, namely griseofulvin and terbinafine, have been documented as inducing BP. These agents share no similarities in their mechanism of action, nor in their molecular structure, and importantly neither agent comprises of a sulfhydryl or phenyl group. Several other agents (actinomycin D, rifampicin, benzyl benzoate) are described in isolated cases with a seemingly unclear association with the succeeding onset of BP. These cases are complicated by several agents being used simultaneously or an uncertain temporal relationship surrounding initiation.
Gliptins. An established body of evidence has accumulated proposing an association of dipeptidyl peptidase 4 inhibitors (DPP4i), oral hypoglycaemic agents often used alongside metformin in treatment diabetes mellitus type 2, with development of BP. This class of medication was identified through an extensive series of case reports that have implicated vildagliptin, sitagliptin, linagliptin, anagliptin and alogliptin as inducing agents. These associations were further examined in two national pharmacovigilance databases (45, 56) and two controlled observational studies (57, 58); the latter were unfortunately underpowered (57, 58). Kridin & Bergman (7) recently performed a retrospective case-control study to examine the use of DPP4i and the occurrence of BP among patients with diabetes demonstrating that vildagliptin, and to a lesser extent, linagliptin were associated with an increased risk of BP. This association with the gliptins was shown to be independent of metformin exposure. An association between gliptin-associated BP and HLA-DQB1*03:01 has been reported in Japanese patients but was not detected in Finnish BP patients with preceding use of gliptins (59, 60).
Whilst considerable progress has been made, the precise pathogenesis underlying the association of DPP4i and BP remains uncertain. DPP4 is a plasminogen receptor expressed on the surface of cells capable of activating plasminogen with the subsequent formation of plasmin, a fundamental serine protease (61). Plasmin is recognised to cleave the immunodominant domain NC16A of BP180 that subsequently can be detected in the skin of the lesion, and the fluid accumulated in the associated blister (62). Izumi et al. (63) proposed that the inhibition of the plasmin formation by DPP4i may be responsible for the inappropriate cleavage of BP180 with theoretical abnormalities in function and antigenicity of the resultant product. Numerous cell types within dermal and epidermal layers, including keratinocytes, are known to express DPP4, which when inhibited by DPP-4 may enhance eosinophil recruitment into the dermis through eotaxin (CCL11) chemokine and other proinflammatory cytokines (64). Other possible mechanisms could include modification of the immune response, alteration of the antigenic properties of the BMZ, or modifying the activity of proteases, such as the highly homologous separase enzyme, resulting in aberrant the processing and/or degradation of the BP180 antigen (65).
Antirheumatics. D-penicillamine and its analogue, tiobutarit, have been implicated in several case reports of DABP. D-penicillamine comprises a distinctive sulfhydryl group hypothesised to be responsible for the underlying pathogenesis of the drug reaction. These thiol drugs are thought to be able to decrease the activity of Treg, enabling the hyperproduction of autoantibodies towards the antigens of the BMZ (2, 31). Even so, an intricate interaction between the proposed mechanisms may precipitate the condition (66). Penicillamine-associated BP appears late in treatment and in a non-dose dependent manner (66, 67). Interestingly, penicillamine-associated bullous dermatoses with combined features of pemphigus and pemphigoid are also described (68).
Tiobutarit possesses two thiol groups. Yamaguchi et al. (69) described the only known case of BP associated with tiobutarit demonstrating recurrence with re-exposure, suggesting a causal relationship.
Biologics. Biologics have been increasingly reported in the literature in numerous case reports, and case series as inducing BP. The reported biologics have included agents targeting various cytokines, interleukins, and other signalling pathways (70–72).
Tumour necrosis factor-α (TNFα) is a multifunctional proinflammatory cytokine involved in a diverse range of systemic diseases. In this regard, antagonists of TNFα have become instituted as an essential treatment in several autoimmune diseases including rheumatoid arthritis, inflammatory bowel disease and psoriasis. BP has been associated with TNFα inhibitors in various case reports (70–72). The agents implicated include a recombinant fusion protein (etanercept), and the monoclonal antibodies (adalimumab, efalizumab, and infliximab) (70–72).
DABP may occur due to the effect of dysregulated TNFα levels in a previously disrupted immune system resulting from the antecedent disease processes, or alternatively this dysregulation may uncover a subclinical autoimmune condition (73). One theory to explain this apparent disconnect may be that the ability of these agents to induce or treat autoimmune disorders is interrelated with the immunological profile, in particular the levels of IL-4 and IFN-γ (73).
In BP, mast cells are responsible for release of TNFα and numerous other mediators upon degranulation, which appears be necessary for recruitment of neutrophils and eosinophils to the surrounding tissue (74). TΝFα is found in higher concentrations in blister fluid than other bullous dermatoses and the circulating levels of this cytokine have been correlated with the severity and number of lesions (75). Lui et al. (73) determined the influence of TNFα on eosinophils concluding these cells are able to regulate the immune response based on the microenvironment. TNFα inhibitors are hypothesised to have the ability to suppress both Th1 and Th2 response in certain disease (73). However, TNFα inhibitors have also been demonstrated to increase autoantibody production (76). Whether this is involved in the pathogenesis of DABP is uncertain, although analogous autoimmune considered have been incited and exacerbated when undergoing treatment with TNFα inhibitors (76).
The IL-2 receptor is known to be expressed in significantly higher amounts in BP, and is correlated with disease activity (77). Aldesleukin (recombinant IL-2) has been suspected as a causative agent for DABP (Table SII)
Patients with BP have been demonstrated to have elevated levels of IL-23 and IL-17 contained in the lesional skin and serum (78). Reports describe ustekinumab, an IL-12/23 antagonist with downstream suppression of the Th1 and IL-17 inflammatory pathway, as achieving control of BP in patients with co-existing psoriasis (79), while paradoxically demonstrated to have been associated with causing the condition in others (80). These findings suggest that immunological state may be altered from Th1 to Th2 dominance with the ensuing release of Th2-associated chemokines (eotaxin and monocyte chemoattractant protein-4) known in the pathogenesis of BP (81). However, numerous studies have reported negligible effect on T-cell response whilst undergoing treatment with ustekinumab (82).
PD1-inhibitors targeting programmed cell death protein-1 (PD-1) and programmed death ligand-1 (PD-L1) pathway are capable of augmenting the intrinsic inhibitory control of the immune system, in particular T cell suppression. While this produces an enhanced response towards the tumour, the non-specific activation of the immune system generates numerous immune-related adverse events (irAEs). PD-1/PD-L1 inhibitor associated dermatological toxicity represents a substantial proportion of all irAEs with a reported incidence of 30–40% (83, 84). These adverse events are often mild, though over 21 cases of DABP have been reported due to nivolumab, pembrolizumab, atezolizumab, and durvalumab (84, 85)) (Table SII).
Epidermal growth factor receptor (EGFR) inhibitor-associated BP has been postulated to be explained by the expression of EGFR in basal keratinocytes (86). The precise mechanism is not known, though it is believed to be through the alteration of antigenic properties of molecules in the lamina lucida, or by decreasing the activity of Treg enabling hyperproduction of autoantibodies towards these antigens (87).
Mammalian target of rapamycin (mTOR) inhibitors (sirolimus, everolimus) has been implicated as a causative agent in two cases of BP in renal transplant recipients (88). BP has previously been described in renal transplant patients due to a graft-related cross reaction with the skin, or dysregulation of the immune system non-specifically inducing production of autoantibodies (42, 89). Further research is required to elucidate whether the mechanism responsible for mTOR inhibitors may be related to an ability to exploit these graft-related mechanisms. BRAF-inhibitors have also been reported to induce a pemphigoid-like eruption (90).
Vaccines. Those vaccines implicated in DABP include the influenza, swine flu, tetanus toxoid and tetracoq, herpes zoster virus, rotavirus and the varying constituents of the hexavalent combined vaccines (Table SII).
Each of the vaccines implicated possesses no recognisable similarities between the structure of the vaccine components and the relevant basement membrane proteins, rendering an antibody-mediated response from the components themselves unlikely (91). The augmentation of the immune response may be triggered by the process of vaccination itself inciting an inflammatory cascade leading to a disruption of BMZ integrity with generation of basement membrane specific antibodies (92).
While remaining a rare occurrence, approximately 110 reports of infantile BP exist in the literature, including 21 following childhood vaccinations. In each case, a short latency period was described from vaccination to clinical manifestation with the majority of cases established within a week (range: 5 h–4 weeks) (Table SII).
Several aspects of the cases support a plausible association including the narrow interval between vaccination and the clinical manifestations, the recurrence of these with subsequent exposure to the vaccination and the higher incidence of vaccine-associated BP in infants (93, 94). However, these hypothetical associations have been challenged.
Baroero et al. (95) emphasises an argument against the existence of a true relationship due to the short latency seen in several of the documented cases including their own, as evidenced by the production of IgG beginning 10–14 days post-immunisations; therefore, many of the observed cases would be considered too short for these autoimmune manifestations to develop. Vaccination is a routine practice in developed countries with the absolute rate of BP remaining low, and relatively rare manifestation in infants despite vaccines’ extensive usage (96).
Neuroleptics and Neurological. At present, a degree of uncertainty surrounds whether neuroleptic drugs are a causative agent of BP or whether the underlying neurological disorder may be responsible. Neurological disorders have been demonstrated in several case-control studies to be associated with a higher risk of developing BP than patients without neurological disorders (97). Jedlickova et al. (98) observed that 45% of patients with BP were affected by a neurological disorder as compared with 18% of a control group with other skin diseases. A plausible hypothesis connecting the derivation of both the nervous system and skin from the neural crest has been proposed (99). Chen et al. (99) provided evidence that an immunogenic BP230 exists in both the human epidermis and brain, further postulating that autoantibodies may be generated following pathogenic changes of neurogenic disorders that lead to exposure of the neural isoforms and resultant cross-reactivity with the antigen in the skin.
Be that as it may, numerous studies have demonstrated a significant association with neuroleptic drugs independent of the underlying disease. A series of prospective case-control studies revealed that when compared to the control group significantly more patients with BP were exposed to neuroleptic agents (35, 36). Patsatsi et al. (26) also observed that neuroleptics were more commonly used in patients with BP in their retrospective analysis. While correlation exists for many agents within the neuroleptic category, no specific drugs have been discerned (36, 100). There have been 10 case reports in the literature that implicate amantadine, doxepin, escitalopram, fluoxetine, flupenthixol, gabapentin, galantamine, levetiracetam, and risperidone (Table SII).
At present, over 90 medications have been associated with inducing BP. While the specific causal relationship in many cases remains to be elucidated, we are beginning to unravel the pathogenesis of the drug-reaction. An appreciation of the medications associated with BP enables clinicians to identify potential cases of DABP earlier and cease the offending medication (Fig. 2). Rechallenging patients to confirm these tentative association of BP with the offending medication remains infeasible due to the ethical and safety concerns. Nonetheless, clinicians should employ a high degree of suspicion for DABP. With contemporary studies continuing to investigate genetic susceptibility, underlying mechanisms and natural history of DABP we are likely to develop a greater understanding of those predisposed to the condition, and the medications that may place the certain groups at risk.
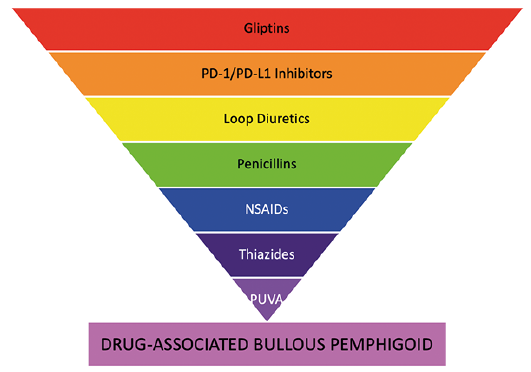
Fig. 2. Diagrammatic summary of strength of evidence derived from systematic review. An inverted pyramid representing the strength of evidence for association with drug-associated bullous pemphigoid (DABP). PD-1: programmed cell death protein-1; PD-L1: programmed death ligand-1; NSAIDS: non-steroidal anti-inflammatory drugs; PUVA: psoralens with UVA.
AB has worked as a Visiting Dermatology Fellow at St. George Hospital, UNSW, Sydney, Australia. She was the recipient of Turkish Society of Dermatology-Prof. Dr. Hulusi Behcet (long-term research) Scholarship in 2018.
The authors have no conflicts of interest to declare.