Apparent Missense Mutation in COL7A1 Causes a Severe Form of Recessive Dystrophic Epidermolysis Bullosa via Effects on Splicing
Syed Ashraf Uddin1,2, Nicole Cesarato2, Aytaj Humbatova2,3, Axel Schmidt2, Fazal ur Rehman4, Muhammad Naeem5, Sabrina Wolf2, Abdul Samad Tareen6, Muhammad Anwar Panezai1, Holger Thiele7, Abdul Wali8, Regina Fölster-Holst9, Sulman Basit10, Muhammad Ayub1# and Regina C. Betz2#
1Institute of Biochemistry, Faculty of Life Sciences, University of Balochistan, Quetta, Pakistan, 2Institute of Human Genetics, University of Bonn, Medical Faculty & University Hospital Bonn, Bonn, Germany, 3Institute of Genetic Resources, Azerbaijan National Academy of Sciences, Baku, Azerbaijan, 4Department of Microbiology, Faculty of Life Sciences, University of Balochistan, Quetta, 5Pakistan Naval Services, Sick Bay, Karsaz, Karachi, 6Post graduate Medical Institute, Quetta, Pakistan, 7Cologne Center for Genomics, University of Cologne, Cologne, Germany, 8Department of Biotechnology, Faculty of Life Sciences, BUITEMS, Quetta, Pakistan, 9Department of Dermatology, University Hospital Kiel, Kiel, Germany and 10Center for Genetics and Inherited Diseases, Taibah University Almadinah, Medina, Kingdom of Saudi Arabia
#Equally contributing last authors.
Dystrophic epidermolysis bullosa is an inherited skin disorder characterized by fragile skin that is prone to blistering. We report here a consanguineous Pakistani family with two siblings, in whom a severe recessive dystrophic epidermolysis bullosa was suspected. Using whole-exome sequencing for one sibling, the homozygous base substitution c.7249C>G in COL7A1 was identified, and could be confirmed in the other sibling by Sanger sequencing. In our exome data, this mutation was annotated as a missense substitution (p.Gln2417Glu), but in silico tools indicated a possible effect on splicing. Using the ExonTrap vector it was
verified that the mutation leads to activation of a cryptic donor splice site, which leads to loss of 26 nucleotides, and a frame-shift event predicted to result in a truncated protein (p.Q2417Sfs*57). The present report describes an apparent COL7A1 missense substitution with an unexpected consequence on splicing that leads to a severe recessive dystrophic epidermolysis bullosa phenotype.
Key words: epidermolysis bullosa; exome sequencing; missense mutation; splicing; COL7A1.
Accepted Sep 7, 2020; Epub ahead of print Sep 14, 2020
Acta Derm Venereol 2020; 100: adv00275.
doi: 10.2340/00015555-3634
Corr: Muhammad Ayub, Institute of Biochemistry, Faculty of Life Sciences, University of Balochistan, Quetta, Postal Code 87300, Pakistan, and Regina C. Betz, Institute of Human Genetics, Medical Faculty & University Hospital Bonn, Venusberg-Campus 1, Building 13, DE-53127 Bonn, Germany. E-mail: ayub_2004@hotmail.com; regina.betz@uni-bonn.de
This study examined the genetic cause of a severe form of the blister-forming disease epidermolysis bullosa in 2 affected siblings from Pakistan. Exchange of a single base on gene copies from both parents was found in the COL7A1 gene, which should affect the protein properties. However, computer programs predicted a so-called splice effect (altered removal of the introns of a gene). This was proved experimentally in cell culture. Thus, the study shows that a new splice site was created in exon 94, which resulted in a truncated protein.
Epidermolysis bullosa (EB) is a rare genodermatosis characterized by a proneness to blistering of the skin. Various subtypes of EB have been described, which vary in terms of clinical manifestation, severity and underlying genetic defect. EB may also present with extracutaneous signs, including oesophageal strictures, muscular dystrophy, nail dystrophy, scarring alopecia, tracheal epithelial erosion, enamel hypoplasia, and corneal erosions. Recently, a consensus reclassification of inherited EB and other skin fragility disorders was published, and classical EB was divided into 4 subgroups: EB simplex (EBS, autosomal dominant and autosomal recessive); junctional EB (JEB, autosomal recessive); dystrophic EB (DEB, exists as both autosomal dominant (DDEB) and autosomal recessive (RDEB)); and Kindler EB (1). To date, approximately 20 genes have been implicated across the EB subtype spectrum (2, 3).
DEB affects both the skin and the mucous membranes, and is characterized mainly by skin blisters, erosions, scars, pseudosyndactyly, mitten hand, mitten foot, and susceptibility to trauma. The phenotype ranges from very mild (isolated nail dystrophy or localized blistering only) to very severe (generalized blistering and failure to thrive). Autosomal recessive DEB (RDEB) is the most severe subtype of DEB, formerly known as Hallopeau-Siemens type (RDEB-HS), in which patients present with generalized lesions; scarring of the hands and feet, leading to fusion of the digits; and severe mucosal involvement (4, 5).
Both DDEB and RDEB are caused by mutations in the COL7A1 gene (e.g. 4, 6–10). COL7A1 encodes type VII collagen, which is the main component of the anchoring fibrils at the dermo–epidermal junction (11, 12). COL7A1 spans 31,132 bps and 118 exons, and is thus one of the largest genes characterized to date (11). The mRNA of COL7A1 is approximately 9?kb in size (12), and is translated into a 350 kDa proα1 (VII) polypeptide (13). The main part of the mature protein consists of a collagenous segment with a Gly–X–Y repeat sequence, which folds into a collagenous triple helical conformation. This large domain is preceded by an amino-terminal non-collagenous (NC-1) domain, which mediates binding of the anchoring fibrils to the basement membrane above and the dermis below. This is followed by a carboxyl-terminal non-collagenous (NC-2) domain, which enables linkage between the collagen homotrimers (14).
RDEB can be caused by diverse types of mutations, i.e. nonsense mutations, splice site mutations, deletions, insertions, ”silent” glycine substitutions within the triple helix and non-glycine substitutions in the triple helix or non-collagenous NC-2 domain (4, 15, 16).
The current report describes a molecular genetic analysis of 2 individuals from a family with suspected severe RDEB from the remote Balochistan region of Pakistan. Due to the clinical variability of EB, the fact that multiple genes have been implicated in its aetiology and the large number of exons in COL7A1, whole exome sequencing (WES) was used to identify the underlying pathogenic variant in this family. A homozygous single base pair substitution (c.7249C>G) in COL7A1 was identified in both affected individuals. The mutation identified in the present study leads to aberrant splicing, followed by the deletion of 26 nucleotides and premature truncation of the protein (p.Q2417Sfs*57).
Subjects
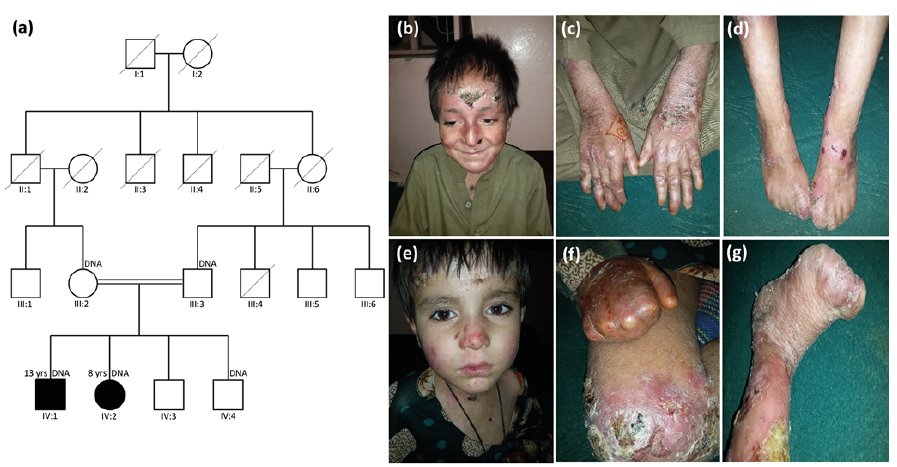
A consanguineous family from Balochistan, Pakistan was investigated. The family comprised 2 siblings, a boy and a girl, aged 13 and 8 years, respectively, with suspected severe RDEB, two unaffected brothers, and their parents (Fig. 1a). The family was interviewed at home. Information concerning the pedigree was obtained from the parents and confirmed via discussions with other relatives.
DNA extraction
All study procedures were performed in accordance with the principles of the Declaration of Helsinki. Ethics approval was obtained from the ethics review committee of the University of Balochistan. Written informed consent was obtained from both parents prior to inclusion. EDTA blood samples were drawn from 5 family members (Fig. 1a), and genomic DNA was extracted via the QIAmp DNA Blood Midi Kit (QIAGEN, Hilden, Germany). DNA purity was determined with a spectrophotometer, and DNA visualization was performed in an ethidium bromide stained 1% agarose gel.
Whole exome sequencing (WES)
For the affected individual IV:2 (Fig. 1a), exome sequencing was performed at the Cologne Center for Genomics (CCG, Cologne, Germany). Details of the sequencing protocol are provided else-where (17, 18). Briefly, 1 μg fragmented DNA was sonicated, and the fragments were subjected to end-repair and adaptor-ligation, including incorporation of sample index barcodes. After size selection, the libraries were enriched using the SeqCap EZ Human Exome Library version 2.0 kit (Roche NimbleGen, Inc., Pleasanton, California, USA). The libraries were then sequenced using a paired end protocol on an Illumina HiSeq 2000 (Illumina, San Diego, CA, USA) instrument. Data filtering and analysis were performed using the Varbank pipeline v.2.18 (https://varbank.ccg.uni-koeln.de/).
Variants were also filtered against the public databases dbSNP, the 1000 Genomes Project, and ExAC. The online tools VariantTaster, PolyPhen2, SIFT, and VariantAssessor were used to predict the pathogenicity of the identified variants.
Sanger sequencing and segregation analysis
For PCR amplification of COL7A1, self-designed primers were used. These primers are obtainable on request. To verify the WES result, Sanger sequencing of the affected individuals and their family members was conducted using the BigDye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems) and an ABI 3100 genetic analyser (Applied Biosystems). Variant location was determined using Seqman software and comparisons with the wild-type sequence, which was extracted from the UCSC Genome Browser website (http://genome.ucsc.edu) (19).
In silico prediction of splice effects
HSF Pro System (https://www.genomnis.com/access-hsf) (20) and SpliceAI (21) were used to analyse the effect of the point variant on splicing. SpliceAI scores were retrieved from the Illumina BaseSpace server on 16 June 2019.
Cloning, cell culture and cDNA preparation
To analyse the effects of the splice site variant (c.7249C>G) on the splicing mechanism in vitro, the wild-type and the mutated sequences were cloned into the Exon trap Cloning Vector (MoBiTec, Göttingen, Germany) using theIn-Fusion® HD Cloning Plus kit (Takara Bio USA, Mountain View, CA, USA) (22). This vector includes an intron that contains a multiple cloning site, which is surrounded by 2 mammalian exons. After transformation of E. coli, plasmids were isolated and correct clones were identified via direct sequencing. Clones containing the desired insert were used for transient transfection of HEK293T cells. Following the extraction of total RNA, reverse transcription was performed. The cDNA obtained was then am-plified and sequenced.
Clinical features of the two affected siblings
Clinically, both affected siblings presented with multiple erosions, atrophic scars, partial ulcers, and crusts and scales on the skin of the face and limbs (Fig. 1b–g). Milia were also observed. Both of the siblings presented with bilateral pseudosyndactyly. The older sibling (IV:1) had partial and complete adhesions between the fingers and toes, respectively (Fig. 1c, d). In the younger sibling (IV:2), the digits of hands and feet showed severe con-tractures with a cocoon-like covering (mitten hand) (Fig. 1f, g). She also presented with oesophageal stenosis and mucosal involvement on the lower lip.
Exome data analysis identified a novel variant in the COL7A1 gene
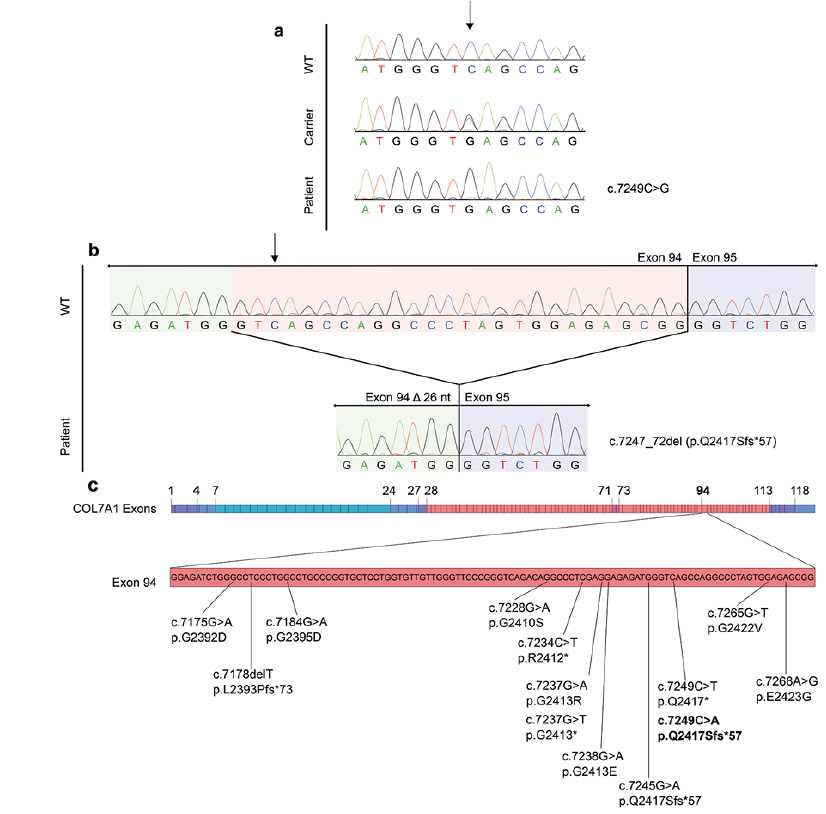
Analysis of the WES data revealed a homozygous base change c.7249C>G in COL7A1. The mutation was veri-fied by Sanger sequencing. Affected and unaffected family members marked in the pedigree (Fig. 1a) were examined for this mutation. Both affected individuals (IV:1, IV:2) were homozygous, while both parents (III:2, III:3) were heterozygous (Fig. 2a). The unaffected sibling (IV:3) carried the wild-type sequence (Fig. 2a). This revealed that in the present family, the variant co-segregated with the disease.
The identified mutation is not present in ExAC or the 1000G database (http://exac. broadinstitute.org/). This mutation is predicted to result in the substitution of the conserved amino acid glutamine by glutamate (p.Gln2417Glu). The in silico tool VariantTaster predicted that the mutation is “possibly damaging”, and PolyPhen2 predicted “disease causing”. However, SIFT predicted that the mutation is tolerated.
In silico analysis predicted activation of a cryptic splice site
HSF Pro predicted activation of a cryptic donor site and a potential alteration of the splicing as a consequence of the single base pair substitution (c.7249C>G) in exon 94. More specifically, the score of the cryptic splice site varied by 191.53% (2.48–7.23) upon substitution. SpliceAI predicted that the mutation decreases by 0.65 the probability that the canonical donor site is used.
Exon Trap studies revealed that the mutation causes the deletion of 26 nucleotides
Due to the lack of RNA samples from the siblings, the splicing effect of the mutation was analysed via the Exon Trap vector (23). Sequencing of the cDNA confirmed the prediction of HSF Pro, as it revealed the creation of a new donor site in exon 94, with the subsequent deletion of 26 nucleotides (Fig. 2b). This altered splicing event shifts the reading frame of COL7A1 mRNA, leading to a stretch of 56 abnormal residues and a premature stop codon that is predicted to result in the truncated protein p.Q2417Sfs*57.
This study identified a causal homozygous single base pair substitution in the exonic region of the gene COL7A1 in 2 Pakistani siblings with suspected severe RDEB. This mutation was predicted to result in the substitution of glutamine by glutamate (p.Gln2417Glu). However, in silico splice predictions indicated a possible effect on splicing. Using an exon-trapping vector, we validated the splicing effect, which indicates a loss of function for COL7A1.
Defects in mRNA splicing are a frequent cause of Mendelian disorders (23). Splicing is dependent on the correct recognition of 3’ and 5’ splice acceptor and donor sites by the splicing machinery (spliceosome). Mutations in the exonic sequences can affect splicing according to 2 different mechanisms: (i) they can create a new 5´ or 3´ splice site, impair regular (canonical) ones or activate cryptic splice sites; (ii) they can disrupt or introduce cis-acting elements, i.e. exonic splicing enhancer or exonic splicing silencer motifs (24–26). These mutations could lead to intron retention or exon skipping (entirely or partially), with the resulting sequence being either in or out of frame. In our case, an exonic C to G substitution in COL17A leads to activation of a cryptic donor site by increasing its conformity to the consensus sequence (strengthening of the cryptic splice site). The mutation leads to recognition of the adjacent GT dinucleotide motif as the 5’ end of an intron by the spliceosome. In the literature, 3 other apparent missense mutations in COL7A1 were reported to have consequences on splicing (Fig. 2c). In either of these cases, a certain amount of wild-type transcript could be detected using skin biopsy samples, and the patients were reported to have mild to intermediate RDEB phenotypes (27–29). Intriguingly, in one of these studies (28), the identified mutation (c.7245G>A) is located just 4 bps upstream of our mutation (Fig. 2c) and leads to the activation of the same cryptic splice site, thus generating the identical aberrant transcript encoding for p.Q2417Sfs*57. In detail, c.7245G>A leads to a substitution that is 2 bps upstream of the GT dinucleotide motif, and also increases the conformity of this cryptic splice site to the consensus sequence. We performed an in silico comparison of the 2 mutations using HSF Pro, which suggested a stronger effect for the mutation observed in the Pakistani siblings. More specifically, while the current mutation led to a 191% score enhancement (as indicator of the increase in likelihood that a splicing event occurs at this location (30)), the mutation reported by Titeux et al. (28) led to a score enhancement of 93.95% (from 2.48 to 4.81). Of note, c.7245G>A was detected in hemizygosity (the second allele was deleted), but the authors could identify a minor amount of wild-type transcript in cultured cells obtained from skin biopsies. Immunostaining in histological skin biopsy sections also revealed a severely reduced but positive signal for collagen VII, confirming residual usage of canonical splice site. Accordingly, Titeux et al. (28) suggested that this may be the reason for the manifestation of an intermediate rather than a severe phenotype. Based on the comparative in silico evaluation it may be plausible that our mutation is associated with an even further decreased usage of the canonical splice site and thus production of even less wild-type protein. This may be the underlying reason for the observed severe RDEB phenotypes in the Pakistani siblings.
However, we must make a cautious remark here regarding the distinction between intermediate and severe RDEB. There is a large overlap in clinical presentation of these 2 forms (31) and to make a clear distinction is challenging at times. Taken that this has a prognostic value (32), ascertainment of better defined criteria for a reliable distinction may aid in this task.
The present analyses did not exclude the possibility that, in tissues in which tissue-specific splicing factors are expressed, the mutation could lead to a different splicing pattern (33).
Severe forms of RDEB are generally caused by nonsense, frameshift or splice-site variants, and in some cases by missense mutations in specific domains (4, 6, 13). Thus, the present experimental findings are consistent with both the severe phenotype of the 2 affected siblings, as well as with the previously described mutations for severe RDEB.
In summary, this study demonstrated that an exonic single-base substitution, initially annotated as a missense mutation, in fact activates a cryptic donor splice site located in exon 94 of COL7A1. Altered splicing results in a frameshift in the transcript and a premature stop codon. The data presented here highlight that single nucleotide changes, with presumably simple effects (silent or missense substitutions), could have rather unexpected consequences on splicing that can be revealed upon closer inspection. Interestingly, 3 other studies reported similar effects for 3 respective COL7A1 missense mutations. Based on their and our findings, a plausible hypothesis is that some of the previously identified single nucleotide changes in COL7A1 may have been misclassified as missense mutations. Furthermore, when interpreted together, the findings of this study and others implicate that the extent of altered splice site usage could have relevance for the severity grade of the resulting RDEB phenotype. These aspects await elucidation by phenotypic and molecular genetic investigations in a larger cohort of patients carrying COL7A1 missense mutations.
The authors thank all family members for their participation.
The data that support the findings of this study are available on request from the corresponding author.
This work was supported by grant No. 20-2407/R&D/NRPU/HEC/11 from the Higher Education Commission of Pakistan (to SAU); the Merit Scholarship Programme for High Technology from the Islamic Development Bank (to AH); by a grant from the GIF, the German-Israeli Foundation for Scientific Research and Development (to RCB); and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), under the auspices of the Germany Excellence Strategy – EXC2151-390873048 (to RCB).
- Has C, Liu L, Bolling MC, Charlesworth AV, El Hachem M, Escámez MJ, et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol 2020; 182: 574–592.
View article Google Scholar - Has C, Nyström A, Saeidian AH, Bruckner-Tuderman L, Uitto J. Epidermolysis bullosa: Molecular pathology of connective tissue components in the cutaneous basement membrane zone. Matrix Biol 2018; 71–72: 313–329.
View article Google Scholar - McGrath JA. Recently identified forms of epidermolysis bullosa. Ann Dermatol 2015; 27: 658–666.
View article Google Scholar - Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol 2008; 17: 553–568.
View article Google Scholar - Koshida S, Tsukamura A, Yanagi T, Nakahara S, Takeuchi Y, Kato T, et al. Hallopeau-Siemens dystrophic epidermolysis bullosa due to homozygous 5818delC mutation in the COL7A gene. Pediatr Int 2013; 55: 234–237.
View article Google Scholar - Uitto J, Pulkkinen L, Christiano AM. Molecular basis of the dystrophic and junctional forms of epidermolysis bullosa: mutations in the type VII collagen and kalinin (laminin 5) genes. J Invest Dermatol 1994; 103: 39S–46S.
View article Google Scholar - Serafi R, Jelani M, Almramhi MM, Mohamoud HSA, Ahmed S, Alkhiary YM, et al. Identification of two homozygous sequence variants in the COL7A1 gene underlying dystrophic epidermolysis bullosa by whole-exome analysis in a consanguineous family. Ann Hum Genet 2015; 79: 350–356.
View article Google Scholar - Eichstadt S, Tang JY, Solis DC, Siprashvili Z, Marinkovich MP, Whitehead N, et al. From clinical phenotype to genotypic modelling: incidence and prevalence of recessive dystrophic epidermolysis bullosa (RDEB). Clin Cosmet Investig Dermatol 2019; 12: 933–942.
View article Google Scholar - Rehman AU, Peter VG, Quinodoz M, Dawood M, Rivolta C. Whole-exome sequencing in a consanguineous Pakistani family identifies a mutational hotspot in the COL7A1 gene, causing recessive dystrophic epidermolysis bullosa. Clin Dysmorphol 2020; 29: 86–89.
View article Google Scholar - Nakamura E, Majima Y, Hashizume H, Tokura Y, Nakano H. Dominant dystrophic epidermolysis bullosa pruriginosa with a COL7A1 exon 87 c.6898CT mutation. Clin Exp Dermatol 2019; 44: 82–84.
View article Google Scholar - Christiano AM, Hoffman GG, Chung-Honet LC, Lee S, Cheng W, Uitto J, et al. Structural organization of the human type VII collagen gene (COL7A1), composed of more exons than any previously characterized gene. Genomics 1994; 21: 169–179.
View article Google Scholar - Christiano AM, Greenspan DS, Lee S, Uitto J. Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. J Biol Chem 1994; 269: 20256–20262.
View article Google Scholar - Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet 2007; 44: 181–192.
View article Google Scholar - Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol 1993; 101: 252–255.
View article Google Scholar - Hamada T, Fukuda S, Ishii N, Sakaguchi S, Ishikawa T, Abe T, et al. Genotype-phenotype correlation in non-Hallopeau-Siemens recessive dystrophic epidermolysis bullosa: the splice site mutation c.6216+5G T in the COL7A1 gene results in aberrant and normal splicings. J Dermatol Sci 2008; 50: 147–150.
View article Google Scholar - Park J, Chae H, Kim M, Kim Y, Park IY, Shin JC, et al. A novel COL7A1 mutation in a Korean patient with Hallopeau-Siemens recessive dystrophic epidermolysis bullosa. Genet Mol Res 2013; 12: 678–682.
View article Google Scholar - Almatrafi A, Alfadhli F, Khan YN, Afzal S, Hashmi JA, Ullah A, et al. A heterozygous mutation in the triple helical region of the Alpha 1 (II) chain of the COL2A1 protein causes non-lethal spondyloepiphyseal dysplasia congenita. Genet Test Mol Biomarkers 2019; 23: 310–315.
View article Google Scholar - Alharby E, Albalawi AM, Nasir A, Alhijji SA, Mahmood A, Ramzan K, et al. A homozygous potentially pathogenic variant in the PAXBP1 gene in a large family with global developmental delay and myopathic hypotonia. Clin Genet 2017; 92: 579–586.
View article Google Scholar - Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res 2002; 12: 996–1006.
View article Google Scholar - Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37: e67.
View article Google Scholar - Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell 2019; 176: 535–548.e24.
View article Google Scholar - Duyk GM, Kim SW, Myers RM, Cox DR. Exon trapping: a genetic screen to identify candidate transcribed sequences in cloned mammalian genomic DNA. Proc Natl Acad Sci U S A 1990; 87: 8995–8999.
View article Google Scholar - Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 2017; 136: 665–677.
View article Google Scholar - Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet 2018; 59: 253–268.
View article Google Scholar - Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002; 3: 285–298.
View article Google Scholar - Blencowe BJ. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci 2000; 25: 106–110.
View article Google Scholar - Gardella R, Zoppi N, Zambruno G, Barlati S, Colombi M. Different phenotypes in recessive dystrophic epidermolysis bullosa patients sharing the same mutation in compound heterozygosity with two novel mutations in the type VII collagen gene. Br J Dermatol 2002; 147: 450–457.
View article Google Scholar - Titeux M, Mejía JE, Mejlumian L, Bourthoumieu S, Mirval S, Tonasso L, et al. Recessive dystrophic epidermolysis bullosa caused by COL7A1 hemizygosity and a missense mutation with complex effects on splicing. Hum Mutat 2006; 27: 291–292.
View article Google Scholar - Wessagowit V, Kim S-C, Woong Oh S, McGrath JA. Genotype-phenotype correlation in recessive dystrophic epidermolysis bullosa: when missense doesn’t make sense. J Invest Dermatol 2005; 124: 863–866.
View article Google Scholar - Jian X, Boerwinkle E, Liu X. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet Med 2014; 16: 497–503.
View article Google Scholar - Fine J-D, Bruckner-Tuderman L, Eady RAJ, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70: 1103–1126.
View article Google Scholar - Shinkuma S. Dystrophic epidermolysis bullosa: a review. Clin Cosmet Investig Dermatol 2015; 8: 275–284.
View article Google Scholar - Grosso AR, Gomes AQ, Barbosa-Morais NL, Caldeira S, Thorne NP, Grech G, et al. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res 2008; 36: 4823–4832.
View article Google Scholar