ORIGINAL REPORT
Sweet Syndrome: Clinical Presentation, Malignancy Association, Autoinflammatory Disorders and Treatment Response in a Cohort of 93 Patients with Long-term Follow-up
Javier GIL-LIANES, Mar LUQUE-LUNA, Francesc ALAMON-REIG, Xavier BOSCH-AMATE, Laura SERRA-GARCÍA and José M. MASCARÓ Jr
Department of Dermatology, Hospital Clínic de Barcelona, Universitat de Barcelona, Barcelona, Spain
Sweet syndrome is a neutrophilic dermatosis associated with multiple disorders. This retrospective case-series study of patients with Sweet syndrome in a tertiary hospital in Spain from 2001 to 2021, explores clinicopathological characteristics of Sweet syndrome and variables associated with malignancy, presence of autoinflammatory disorders and differences between histological subtypes. A total of 93 patients were identified: 30% idiopathic, 34% malignancy-associated, 29% reactive to infections or drug-associated, and 6% with an autoimmune/inflammatory condition. Acute myeloid leukaemia was the most common malignancy (16/93) followed by myelodysplastic syndrome (7/93). Patients with acute myeloid leukaemia presented isolated flares, marked cytopaenia and rapid response to treatment, whereas myelodysplastic syndrome followed a chronic-recurrent course. The most frequent associated medications and inflammatory disorders were filgrastim and hydroxyurea (n = 2); and inflammatory bowel disease (n = 4). In addition, 3 patients were diagnosed with VEXAS syndrome. Male sex (p = 0.006), fever (p = 0.034), increased erythrocyte sedimentation rate (p < 0.001), anaemia (p < 0.001), and thrombocytopaenia (p < 0.001) were associated with malignancy. Histologically, patients were classified as classic (60%), histiocytoid (22.5%) or subcutaneous (15%), with pain (p = 0.011) and nodules (p < 0.001) being associated with subcutaneous-Sweet syndrome. Sweet syndrome in the context of cytopaenia should alert the presence of malignancy. An acquired autoinflammatory condition should be explored in relapsing Sweet syndrome with myelodysplastic syndrome. A minimum follow-up of 6 months is recommended.
SIGNIFICANCE
Sweet syndrome is an inflammatory skin disorder linked to various diseases, such as haematological malignancies. In this study, 93 patients with Sweet syndrome were examined. Acute myeloid leukaemia and myelodysplastic syndrome were the most common malignancies. In addition, 3 patients with myelodysplastic syndrome had a VEXAS syndrome diagnosed, which is a recently described autoinflammatory condition with poor prognosis. Medications, such as filgrastim and hydroxyurea, and inflammatory bowel disease were also frequently encountered. Males with fever, increased erythrocyte sedimentation rate, anaemia, and thrombocytopaenia were at risk for malignancy. Taking these factors into account is key to direct the study and treatment of patients with Sweet syndrome.
Key words: acute febrile neutrophilic dermatosis; acute myeloid leukaemia; autoinflammatory syndrome; neutrophilic dermatosis; Sweet syndrome; VEXAS syndrome.
Citation: Acta Derm Venereol 2023; 103: adv18284. DOI: https://doi.org/10.2340/actadv.v103.18284.
Copyright: © Published by Medical Journals Sweden, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/)
Accepted: Oct 3, 2023; Published: Dec 19, 2023
Corr: José M. Mascaró Jr, Department of Dermatology, Hospital Clínic de Barcelona, Universitat de Barcelona, Calle Villarroel, 170, ES-08036 Barcelona, Spain. E-mail: jmmascaro_galy@ub.edu
Competing interests and funding: JMM has received speaker fees from Academia Española de Dermatología, Argenx BV, Bocemtium Consulting, Clover Soluciones Globales de Marketing, Ferrer Internacional, Fundació Clínic per la Recerca Biomédica, LEO Pharma Spain, Loki & Dimas, Luzan 5 Health Consulting, and M.S.D. de España S.A, S&H Medical Science Service, and Sanofi-Aventis all outside the submitted work.
INTRODUCTION
Sweet syndrome (SS), or acute febrile neutrophilic dermatosis, is an inflammatory disorder characterized by fever and tender papules, plaques or nodules, with a histology characterized by a dense neutrophilic infiltrate in the reticular dermis (1–3) and oedema, without primary vasculitis. Since its first description in 1964 (4), Sweet syndrome has been associated with multiple conditions, which have been included in its diagnostic criteria (1, 5) and highlighted the importance of categorization into further subtypes (idiopathic, reactive, malignancy-associated). Recently, Sweet syndrome has been classified within the spectrum of autoinflammatory disorders, although its precise aetiology remains elusive (2, 6). Additional histopathological subtypes have been described, namely histiocytoid-SS (7, 8) and subcutaneous-SS (9, 10), whose clinical implications are still controversial. To date, studies including large cohorts of this rare condition are scarce with minimal follow-up.
The aims of this study were to describe the clinicopathological characteristics of SS in a large cohort of 93 patients with a long-term follow-up, determine possible indicators of malignancy, explore the presence of autoinflammatory disorders and describe differences between histopathological subtypes.
MATERIALS AND METHODS
Study design, setting and participants
A retrospective study was conducted to identify patients with SS from a tertiary referral centre in Spain between 2001 and 2021. Two physicians independently examined medical records to confirm patients met Su and Liu diagnostic criteria (1). Clinicopathological data were obtained from medical records.
Patients were categorized into 4 groups according to their clinical presentation: malignancy-associated (MA-SS), reactive-SS (11), inflammatory/autoimmune-associated (A-SS) and idiopathic-SS. Patients were defined as MA-SS if they satisfied the Curth postulates (12) and as reactive-SS if their presentation occurred in the setting of a previous infection with a plausible chronological relation or was drug-associated according to the modified Walker and Cohen diagnostic criteria (11). Patients with a systemic inflammatory or autoimmune condition, without other identifiable factors, were classified as A-SS. If patients could not be characterized as belonging to these groups due to the absence of a predisposing factor, they were classified as idiopathic-SS.
Histopathological subtypes were classified into classic-SS, subcutaneous-SS (9, 10, 13) and histiocytoid-SS (7, 8). Further specifications of data collection and patient classification are detailed in Appendix S1.
Statistical analysis
Data were analysed with Stata software (v17.0, StataCorp LLC, Texas, USA). Continuous variables were summarized with means and standard deviations (SD) and compared using Student’s t-test or analysis of variance (ANOVA) for normal data distribution. Categorical variables were reported as proportions or percentages, and comparisons between groups were performed using the χ2 test. p-values < 0.05 were considered statistically significant. Regression analyses were also performed to identify predictor variables for MA-SS.
RESULTS
Patient characteristics
Of the 207 patients with a possible diagnosis of SS, 93 fulfilled diagnostic criteria (1) (Table SI). Their clinicopathological characteristics are summarized in Table I. Mean age of presentation was 59.3 years and 50.5% were female. Papules or plaques were the predominant morphology (65.6%), followed by nodules (15%), vesicle-bullous (7.5%), or pustules (5.4%), although different types could coexist (Fig. 1). Patients usually had more than 1 affected region, chiefly the upper extremities and trunk. Most patients (88.1%) had additional symptoms, primarily fever (68.8%) and pain (61.2%).
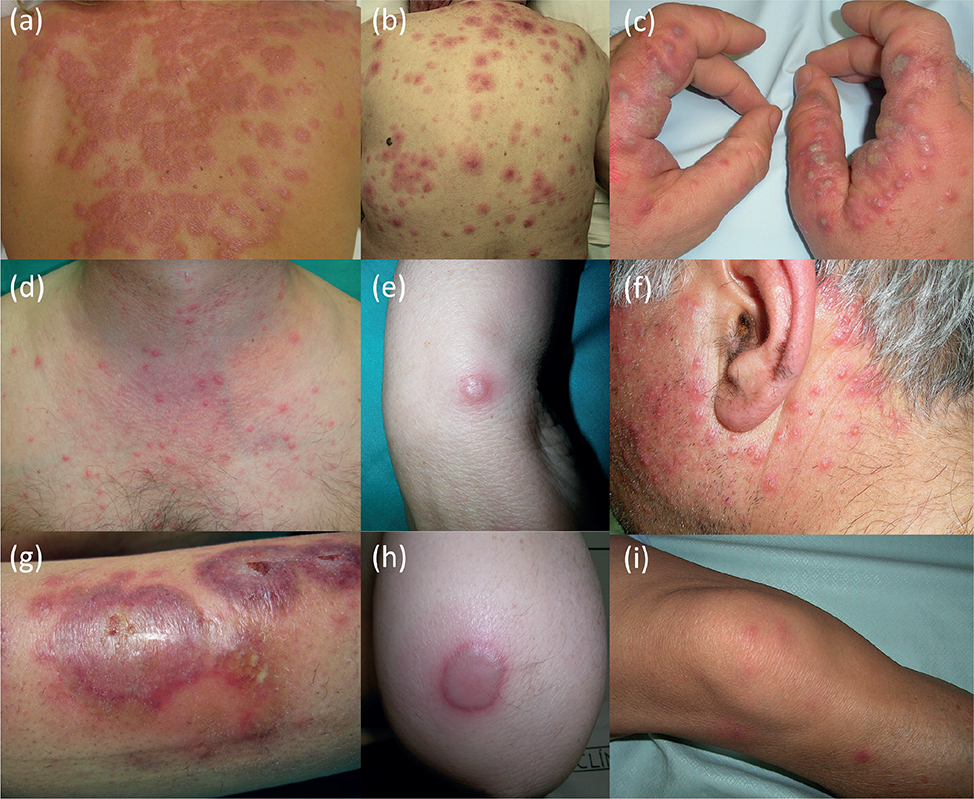
Fig. 1. Clinical presentation of Sweet syndrome (SS). The main types of lesions found in this study are shown here. Of note is the finding that different morphologies can coexist in the same patient (g). (a, b) Erythematous, oedematous, and infiltrated plaques on the trunk. (c, f) Grouped and pleomorphic pustules, with an erythematous base, affecting (c) the hands and (f) the face. (d) Indurated, oedematous, non-grouped, erythematous papules, asymmetrically distributed, in the upper anterior trunk. (e, h) Isolated, seropurulent bullous lesions on the elbow. (g) Pleomorphic lesions consisting of atypical targetoid plaques, with eroded bullous central lesions and peripheral pustular lesions on the leg. (i) Indurated, ill-defined, erythematous nodules asymmetrically distributed on the legs. (b) and (d) are from 2 patients with VEXAS syndrome.
Of the 86 patients with laboratory analysis, 29 (33.7%) had 3 or more abnormal values included in the Sweet syndrome diagnostic criteria (1). Increased levels of C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) were found in 87.3% and 92.5% of patients, and a third had leukocytosis (32.9%) or neutrophilia (33.3%). Anaemia was found in 64.6% of patients (mean haemoglobin level, 11.1 g/dL), macrocytosis in 10.9%, and thrombocytopaenia in 29.4%. Liver enzymes were increased in 13.9% of patients, none above 3 times the normal value.
Associated conditions and medications
The study found 65 patients (69.9%) with a potentially associated condition at diagnosis. Infections, medication, inflammatory disorders, and malignancies were present in 23.6%, 28.2%, 9.7% and 40.9% of patients. Factors such as myelodysplastic syndrome and VEXAS syndrome or neutropaenic patients, invasive fungal infections and their treatment coexisted as potential triggers. Individual cases were studied and the most plausible trigger chosen. The associated factors classified patients into 4 groups: idiopathic-SS (n = 28, 30.1%), MA-SS (n = 32, 34.4%), reactive-SS (n = 27, 29%) or A-SS (n = 6, 6.4%).
Autoimmune/inflammatory disorders were present in 6 patients: inflammatory bowel diseases (IBD), with Crohn’s disease (n = 2) and ulcerative colitis (n = 2). Other inflammatory conditions were hidradenitis suppurativa (n = 1) and cryopyrin-associated autoinflammatory syndrome (n = 1). Infections were present in 18 patients, where 55.6% were bacterial infections, followed by viral infections (38.8%) and parasitic infections (5.6%). Twenty-six patients were under a potentially associated medication at diagnosis, with 9 drugs satisfying the Walker and Cohen diagnostic criteria for drug-induced SS, although some occurred in patients with an underlying malignancy (filgrastim and hydroxyurea; n = 2, each). Other associated drugs were SARS-CoV-2 (COVID-19) vaccine, lenalidomide, vemurafenib/trametinib, all-trans retinoic acid and imatinib (n = 1, each).
Thirty-eight patients had a history of active malignancy at diagnosis with 32 satisfying Curth’s postulates (12) for MA-SS. Haematological malignancies were the predominant type, mainly acute myeloid leukaemia (AML, n = 16) and myelodysplastic syndrome (MDS, n = 7). Despite the fact that VEXAS syndrome (n = 3) is an autoinflammatory disorder, it was initially classified in the malignancy-group, as all patients had a MDS. The 2 associated solid neoplasms were a ductal breast carcinoma and a serous ovarian adenocarcinoma, both metastatic.
Clinicopathological features of patients with and without malignancy-associated Sweet syndrome
Table I compares clinicopathological data for patients with and without MA-SS. Male sex (p = 0.006, OR 3.75), fever (p = 0.034, OR 3.46), anaemia (p < 0.001, OR 26), thrombocytopaenia (p < 0.001, OR 19.4), absence of neutrophilia (p = 0.012) and increased ESR levels (p < 0.001) were significantly associated with MA-SS. There were no statistically significant differences in age, other accompanying symptoms, type of lesions, treatment response or relapses.
As for the histopathological findings, classic-SS was the predominant subtype in both groups (MA-SS 46.2%; non-MA-SS 65.4%). Histiocytoid and subcutaneous variants were more frequent in MA-SS (histiocytoid-SS, 30.8% vs 1 11.5%), but without statistically significant differences 9.2%, and subcutaneous-SS, 23.1% vs (p = 0.172).
Clinical features of patients with Sweet syndrome with acute myeloid leukaemia or myelodysplastic syndrome
Table II compares clinicopathological features of patients with AML (n = 16) and MDS (n = 7) to assess its clinical behaviour. AML and MDS differed significantly (p < 0.05) in terms of treatment response, duration of treatment, presence of relapses and level of platelets, although anaemia and leukopaenia were more prevalent in the AML group. SS in patients with AML presented mainly as isolated flares (recurrent-SS, 18.8%), with marked cytopaenia and rapid response to treatment. On the other hand, MDS-SS presented a chronic course (recurrent-SS, 57.1%) with a long duration of treatment (median, 423 days) and increased mean corpuscular volume (median, 101 fl).
| Number of patients | AML | MDS | p-value |
| (n = 16) | (n = 7) | ||
| Age of onset, years, median (range) | 63.0 (52.2–74.8) | 66.0 (54.7–73.5) | 0.923 |
| Sex, male, n (%) | 10, 62.5 | 6, 90 | 0.232 |
| Presence of additional symptoms, n (%) | 15, 93.4 | 7, 100 | 0.452 |
| Time before onset, days, median (range) | 30 (0–530) | 105.0 (0–782) | 0.843 |
| Previous infection < 14 days, n (%) | 3, 18.8 | 2, 30 | 0.392 |
| Drug related < 14 days, n (%) | 3, 18.8 | 0 | 0.272 |
| Treatment response, CR, n (%) | 14, 87.5 | 1, 14.3 | 0.012 |
| Duration of treatment, days, median (range) | 30 (20–70) | 423 (185–782) | 0.023 |
| Relapses of SS, n (%) | 3, 18.8 | 4, 57.1 | 0.022 |
| Number of relapses, n (%) | 0.012 | ||
| 0 | 13, 81.2 | 3, 42.9 | |
| 2 | 3, 18.8 | 0 | |
| > 2 | 0 | 4, 57.1 | |
| CRP, mg/dL, median | 10.1 | 11.7 | 0.893 |
| ESR, mm/h, median (range) | 104.2 | 113.3 | 0.643 |
| Haemoglobin level, g/dL, median | 8.6 | 9.7 | 0.073 |
| MCV (median, range, fl) | 92.9 | 101 | 0.033 |
| Platelets, × 109/L, median | 24.2 | 92.1 | 0.013 |
| Leukocyte count, × 106/L, median | 720.8 | 5270.5 | 0.143 |
| Neutrophil count, × 106/L, median | 100.4 | 2110.1 | 0.103 |
| 1Kruskal-Wallis test; 2Pearson correlation coefficient; 3Wilcoxon test. AML: acute myeloid leukaemia; CR: complete response; ESR: erythrocyte sedimentation rate; MCV: mean corpuscular volume; MDS: myelodysplastic syndrome; CRP: C-reactive protein; SS: Sweet syndrome. | |||
Molecular findings in patients with acute myeloid leukaemia
Clinical, cytogenetic, and targeted next-generation sequencing (NGS, Table SII) data were obtained for 16 patients with associated AML and are summarized in Table III. Fourteen patients (87.5%) had de novo AML, and 2 (12.5%) had AML arising from MDS. Thirteen patients (81.3%) had active disease at diagnosis and 3 (18.8%) were in remission. In 7 patients (43.75%) SS and AML diagnosis was simultaneous. Three patients (18.8%) had relapses concurrent with AML relapses. Notably, SS occurred in the presence of other concomitant factors: 3 patients had had a previous infection (2 bacterial, 1 fungal) and 2 patients were being treated with filgrastim.
| Characteristic | N = 16 | N | % |
| Classification | De novo | 14 | 87.5 |
| Arising from myelodysplastic syndrome | 2 | 12.5 | |
| Karyotype | |||
| Favourable | 0 | ||
| Intermediate | Normal, Other karyotypes not included in the other 2 groups | 11 | 68.75 |
| Adverse | Complex*, Inv(3)(q21q26), del(7q) | 4 | 25 |
| Unknown | 1 | 6.25 | |
| Targeted next-generation sequencing | N = 13 | % | |
| Number of disease-associated mutation, median (range) | 3 (1–5) | ||
| Specific mutations** | |||
| ASXL1 | 2 | 15.38 | |
| BCOR | 1 | 7.69 | |
| DNMT3A | 6 | 46.15 | |
| FLT3 | 4 | 30.77 | |
| IDH1 | 4 | 30.77 | |
| KRAS | 1 | 7.69 | |
| NPM1 | 5 | 38.46 | |
| RUNX1 | 2 | 15.38 | |
| SRSF2 | 1 | 7.69 | |
| TET2 | 1 | 7.69 | |
| WT1 | 1 | 7.69 | |
| *Karyotypes with 4 or more unrelated abnormalities are considered complex as defined by the cytogenetic classification of World Health Organization (WHO) Classification category and prognostic risk grouping. **Mutations are not mutually exclusive; thus, the total percentage exceeds 100%. ASXL1: ASXL transcriptional regulator 1; BCOR: BCL6 corepressor; DNMT3A: DNA methyltransferase 3 alpha gene; FLT3: fms-like tyrosine kinase 3 gene; IDH1: isocitrate dehydrogenase 1 gene; KRAS: Kirsten rat sarcoma virus; NPM1: nucleophosmin 1 gene; RUNX1: runt-related transcription factor 1; SRSF2: Serine and arginine rich splicing factor 2; TET2: tet methylcytosine dioxygenase 2 gene; WT1: Wilms tumor 1 gene. | |||
Of the 16 patients with AML, cytogenetic analysis revealed either intermediate (68.8%) or adverse (25%) karyotypes. In 13 patients, a NGS panel for somatic mutations associated with haematological malignancies was performed. The median number of mutations was 3 (range 1–5), mainly DNMT3A (n = 6), NPM1 (n = 5), FLT3 (n = 4) and IDH1 (n = 4).
Clinical features of different histopathological patterns: classic, subcutaneous and histiocytoid Sweet syndrome
Of the 80 patients for whom we could review histopathological findings, these were classified into 3 groups: classic (n = 48, 60%), histiocytoid (n = 18, 22.5%) and subcutaneous (n = 12, 15%). In addition, 2 patients (2.5%) could not be assigned to a specific group due to the presence of mixed infiltrates.
Table IV compares clinical and analytical findings between classic, histiocytoid and subcutaneous-SS. Subcutaneous-SS was significantly associated with the presence of pain (p = 0.011) and a distinct type of lesion (p < 0.001), as nodular lesions were present in 66.7% of cases. There were no other statistically significant differences. Despite that, MA-SS accounted for 44.2% and 50% of histiocytoid-SS and subcutaneous-SS cases, respectively, but only 27.1% of classic-SS cases (p = 0.383).
| Parameter | Classic | Histiocytoid | Sub-cutaneous | p-value |
| Patients | 48 | 18 | 12 | |
| Age of onset, years, mean | 58 | 58.6 | 64.1 | 0.673 |
| Sex, male, n (%) | 25 (52.1) | 7 (38.9) | 4 (33.3) | 0.542 |
| Aetiology, n (%) | 0.383 | |||
| Idiopathic | 13 (27.1) | 6 (33.3) | 3 (25) | |
| Reactive | 20 (41.7) | 3 (16.7) | 2 (16.7) | |
| Malignancy | 13 (27.1) | 8 (44.4) | 6 (50) | |
| Autoimmune and inflammatory conditions | 2 (4.2) | 1 (5,6) | 1 (8.3) | |
| Type of lesions, n (%) | < 0.001 | |||
| Papule-plaque | 36 (75) | 16 (88.9) | 4 (33.3) | |
| Vesicle-bullous | 4 (8.4) | 0 | 0 | |
| Nodules | 2 (4.2) | 2 (11.1) | 8 (66.7) | |
| Pustules | 3 (6.3) | 0 | 0 | |
| Unknown | 3 (6.3) | 0 | 0 | |
| Additional symptoms, n (%) | 40 (83.3) | 16 (88.9) | 12 (100) | 0.243 |
| Fever | 31 (64.6) | 15 (83.3) | 12 (100) | 0.349 |
| Pain | 29 (60.4) | 8 (44.4) | 12 (100) | 0.011 |
| Arthralgia | 8 (16.7) | 4 (22.2) | 5 (41.7) | 0.083 |
| Weight loss | 5 (10.4) | 5 (27.8) | 0 | 0.061 |
| Ocular/ear | 3 (6.3) | 1 (5.6) | 0 | 0.615 |
| Treatment response, CR/PR, % | 80/20 | 57.14/42.86 | 58.33/41.67 | 0.147 |
| Relapses, n (%) | 14 (29.1) | 6 (33.3) | 3 (25) | 0.887 |
| Laboratory findings | ||||
| CRP >2 mg/dL, % | 92.5 | 86.67 | 83.33 | 0.606 |
| CRP, mg/dL, mean | 10.5 | 11.5 | 11.6 | 0.880 |
| ESR >20 mm/h, % | 94.44 | 90 | 85.71 | 0.769 |
| ESR, mm/h, mean | 73.3 | 86.9 | 62.4 | 0.340 |
| Anaemia, % | 66.67 | 75 | 61.54 | 0.728 |
| Haemoglobin level, g/dL, mean | 11.2 | 10.4 | 10.8 | 0.146 |
| MCV >100 fl, % | 14.58 | 12.5 | 0 | 0.190 |
| MCV, fl, mean | 94.3 | 90.3 | 92.9 | 0.930 |
| Thrombocytopaenia, % | 27.27 | 33.33 | 50 | 0.436 |
| Platelets, ×109/L, mean | 218.8 | 199.5 | 228.4 | 0.330 |
| Leukocytosis, % | 36.36 | 26.67 | 30.77 | 0.771 |
| Leukocyte count, ×106/L, mean | 9,603.6 | 11,718.7 | 5,821.8 | 0.354 |
| Neutrophilia, % | 40.91 | 20 | 30.77 | 0.332 |
| Neutrophil count, ×106/L, mean | 6,001.9 | 12,084.0 | 3,934.5 | 0.347 |
| Elevated AST and/or ALT, % | 15 | 20 | 15.38 | 0.901 |
| aFifteen patients were not included in this analysis. Although we had the histopathological diagnosis of Sweet syndrome, we did not have the specifications of the infiltrate in 13 patients, and two other patients could not be classified into a specific group due to the presence of mixed infiltrates. Thus, we could not distribute these 15 patients in either category. CR: complete response; PR: partial response; ESR: erythrocyte sedimentation rate; MCV: mean corpuscular volume; CRP: C-reactive protein; ALT: alanine aminotransferase; AST: aspartate aminotransferase. | ||||
Treatment
Of the 93 patients, 70 (75%) received systemic corticosteroids and 15 (16.3%) topical corticosteroids, usually in combination. In acute settings, non-steroidal anti-inflammatory drugs (NSAIDs) (15.2%) and antibiotics (11.9%) were also used. Treatment response could be reviewed in 76 of the 93 patients, with a partial (26.7%) or complete (73.3%) response to corticosteroids. At follow-up, 27.2% patients experienced at least 1 relapse with 7 patients having ≥ 10 relapses. Patients with multiple relapses required the addition of immunomodulatory therapies, such as methotrexate (n = 3), supersaturated potassium iodide (n = 4), hydroxychloroquine (n = 2) or cyclosporine (n = 2) (Table SIII).
Follow-up
Mean follow-up was 50.93 months with 69 patients followed up for > 1 year. Four patients developed a haematological malignancy after SS diagnosis: MDS (1 month), an atypical CD30-lymphoproliferative disorder (16 months), large granular lymphocytic leukaemia (36 months) and amyloid light-chain primary amyloidosis (36 months). Six patients developed solid neoplasms (between 34 and 144 months): 2 breast and 2 colon adenocarcinomas, 1 squamous cell lung carcinoma and 1 bladder carcinoma. Of all cases, based on the timeline of events, only 1 with MDS was considered associated and classified as MA-SS. As for other non-malignant conditions: 1 patient had polyarteritis nodosa (24 months) and 1 patient psoriatic arthritis (84 months). In addition, 4 patients were diagnosed a monogenic autoinflammatory condition: 3 patients with VEXAS syndrome and 1 patient with cryopyrin-associated autoinflammatory syndrome (CAPS). During follow-up, the major cause of death was malignancy progression (23 patients, 24.7%), and no deaths were related to SS.
Comparison with other Sweet syndrome series with more than 50 patients
A comparative analysis of clinicopathological features and MA-SS indicators of the study cohort and published series with > 50 patients is shown in Table SIV.
DISCUSSION
Sweet syndrome is a distinctive neutrophilic disorder whose associations and histopathological findings have evolved since Robert Douglas Sweet’s first description (4). To our knowledge, this is one of the largest studies evaluating epidemiological features, malignancy indicators, clinical behaviour of MA-SS and presence of autoinflammatory disorders. This study provides new insights into the characterization of SS.
The mean age of presentation was 59.3 years, similar to the reported peak incidence (50–60 years) (2, 14, 15), with a slight female predominance (50.5%). Historically (16), idiopathic-SS was thought to be more frequent in females (4:1) (2, 6, 17, 18) and a 1:1 relationship in MA-SS (16). However, recent publications have shown a near 1:1 relation overall (14, 15, 19, 20). Leukocytosis and neutrophilia were detected in only one-third of our patients (15, 18). This might be explained by 34.4% of cases being malignancy-associated. Since tissular-neutrophilia in SS can be present in the absence of peripheral-neutrophilia in blood (14), this raises the question of whether peripheral-neutrophilia should be used as a diagnostic criterion in cancer patients in whom it is usually absent.
The distribution of patients between the different aetiological factors was similar to previous reports (14, 15, 17–20) with idiopathic (30.1%) and MA-SS (34.4%) as the most frequent, with MA-SS representing 16–44% in other studies (14–16, 19, 20). Multiple drugs have been associated with SS (2, 6) with all medications in this study having been reported previously, where COVID vaccines are the newest incorporation (21). As for infections, viral and respiratory infections (14, 20) are the most frequently associated, in contrast to our series in which bacterial infections predominate (55.6%). However, isolation of causative pathogens is usually not possible, and type of infection is based on clinical symptoms. The association with autoimmune/inflammatory conditions has been studied extensively, with IBDs (22) being a common finding (23). In this study, 4 patients had IBD.
As for clinicopathological predictors of MA-SS, significant associations were found with male sex, fever, anaemia, thrombocytopaenia, increased ESR and absence of neutrophilia. Although age, vesiculobullous morphology, relapsing-SS, absence of arthralgia and histiocytoid, lymphocytic or subcutaneous histopathology have been described as malignancy indicators in previous studies (2, 6, 14, 15, 18, 19, 24–26), we did not find such associations. Fever was recently described as a risk factor for MA-SS in a systematic review of histiocytoid-SS (27). Nonetheless, fever and MA-SS association might be explained by cancer patients’ vital signs being measured more frequently. Among patients with MA-SS, haematological malignancies were the predominant type (90%), mainly AML (n = 16) and MDS (n = 7), and were usually diagnosed before or at the onset of SS (14, 15, 28). One patient had an MDS diagnosed 1 month after onset of SS. Thus, patients with SS should be monitored and Marcoval et al. (15) have proposed at least 12 months of follow-up. SS incidence in patients with AML is thought to be 1–5.5% (2), with −5/del(5q) karyotype and FLT3 mutations (39% of patients) being more prevalent in patients with AML who develop SS (14, 29–31). In the current study, FLT3 mutations were found in 30.7% of patients, DNMT3A was the most frequent mutation (46.2%), and no patients with deletions in 5q were found. Although MDS and AML represent the major entities of MA-SS, SS behaviour differs between these entities (32, 33). In the current study series, 31% of MA-SS had ≥ 2 relapses. Specifically, of 16 AML-SS, only 3 (18.8%), and of 7 MDS-SS, 4 (57.1%) had ≥ 2 relapses. Thus, MDS-SS presents a chronic-relapsing course and requires long-term corticosteroid therapy or additional therapies, while AML-SS usually presents as a single episode with marked cytopaenia and rapid resolution after corticosteroids (33).
The understanding of SS pathogenesis is incomplete, and it is probably multifactorial. However, certain drugs represent a true offending agent due to immunological modifications (e.g. G-CSF, ATRA, FLT3-inhibitors). Autoinflammation is increasingly thought to play a role in the pathogenesis of neutrophilic dermatoses (2, 34). Recently, VEXAS syndrome, an acquired autoinflammatory disease resulting from a somatic mutation in haematopoietic progenitor cells has been described (35). Common clinical features include neutrophilic dermatoses (36), myelodysplasia with cytoplasmic vacuoles and polychondritis. While the presence of VEXAS syndrome in SS cohorts has not previously been described, in this series we found 3 patients with VEXAS syndrome, all with accompanying MDS. Previous reports of cases of relapsing MDS-SS, polychrondritis (35) and lymphocytic or histiocytoid infiltrates, could, in fact, be VEXAS syndromes. In addition, 1 patient was diagnosed with CAPS due to a pathogenic NLRP3 variant, although neutrophilic urticarial dermatosis is the usual histopathological pattern in CAPS (37).
Histologically, SS was initially defined as a diffuse neutrophilic infiltration of the upper dermis and marked papillary oedema without vasculitis (4). Later, additional histopathological subtypes have been described, such as histiocytoid and subcutaneous-SS (7–10, 19, 27). Of note, different subtypes can coexist, as happened in the current study cases, and could exist as a continuum (2, 38). The current study performed a comparative analysis of classic, subcutaneous and histiocytoid-SS, which has not previously been performed in such a large series. Nodules (p < 0.001) and pain (p = 0.011) being more common in subcutaneous-SS, which can be explained by the typology of the lesion. In addition, despite malignancies were more common in histiocytoid-SS (44.4%) and subcutaneous-SS (50%), compared with classic-SS (27.1%), no significant differences were found (p = 0.383). Histiocytoid-SS association with malignancies has been suggested (7, 8, 19). A large systematic review published in 2020 found patients with histiocytoid-SS to have a higher risk of underlying haematological malignancy compared with those with classic-SS (27), especially if cases are older, recurrent and are accompanied by fever or musculoskeletal symptoms (8, 14, 19). Regarding subcutaneous-SS, it represents approximately 10% of SS cases (2, 9, 24, 39). Recent publications have proposed its association with myeloid malignancies (13, 14, 30, 40), although larger studies are required to avoid misleading information from single case reports.
The retrospective design of this study is a limitation. The study was based at a tertiary academic referral centre, with a leading onco-haematological department, which can limit its generalizability. Therefore, this study should not be used to establish relative epidemiological percentages for malignancy associated, drug-induced, and idiopathic Sweet’s, as the study cohort is biased towards malignancy. Since pathology reports were used to classify the type of histological pattern, some older reports (> 10–15 years old) did not make a distinction between different subtypes of SS, and these patients could not be included in the analysis of different histological subtypes.
In conclusion, this study we found that anaemia, thrombocytopaenia, male sex, fever, increased ESR levels and absence of neutrophilia were significantly associated with MA-SS. Since peripheral-neutrophilia is usually absent in MA-SS, the use of peripheral-neutrophilia as a diagnostic criterion might not be useful in this setting. In MA-SS, onset frequently appeared concomitant or shortly after malignancy diagnosis and AML and MDS remained the most frequent malignancies. It is notable that MDS-SS usually presents a chronic-relapsing course, and this may indicate the presence of autoinflammatory conditions as 3 patients had VEXAS syndrome, which is the first time it has been described in a SS cohort. Future research is necessary to clarify whether neutrophilic dermatoses found in patients with MDS are part of an acquired-autoinflammatory disorder.
ACKNOWLEDGEMENTS
The study has been approved by the Institutional review board: Hospital Clinic of Barcelona in Barcelona (HCB/2021) and guided by the STROBE checklist. This study has not been presented previously. The patients have given written consent for publication and use of clinical photographs and medical information.
REFERENCES
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis 1986; 37: 167–174.
- Joshi TP, Friske SK, Hsiou DA, Duvic M. New practical aspects of Sweet syndrome. Am J Clin Dermatol 2022; 23: 301–318.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol 2003; 42: 761–778.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol 1964; 76: 349–356.
- von den Driesch P, Gomez RS, Kiesewetter F, Hornstein OP. Sweet’s syndrome: clinical spectrum and associated conditions. Cutis 1989; 44: 193–200.
- Nelson CA, Stephen S, Ashchyan HJ, James WD, Micheletti RG, Rosenbach M. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol 2018; 79: 987–1006.
- Requena L, Kutzner H, Palmedo G, Pascual M, Fernández-Herrera J, Fraga J, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol 2005; 141: 834–842.
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, Rodríguez-Peralto JL, Alegre V, Cerroni L, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol 2017; 153: 651–659.
- Chan MP, Duncan LM, Nazarian RM. Subcutaneous Sweet syndrome in the setting of myeloid disorders: a case series and review of the literature. J Am Acad Dermatol 2013; 68: 1006–1015.
- Guhl G, García-Díez A. Subcutaneous Sweet syndrome. Dermatol Clin 2008; 26: 541–551.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol 1996; 34: 918–923.
- Ho C. Skin lesions and internal carcinoma. Cancer Skin 1976; 1308–1341.
- Sutra-Loubet C, Carlotti A, Guillemette J, Wallach D. Neutrophilic panniculitis. J Am Acad Dermatol 2004; 50: 280–285.
- Nelson CA, Noe MH, McMahon CM, Gowda A, Wu B, Ashchyan HJ, et al. Sweet syndrome in patients with and without malignancy: a retrospective analysis of 83 patients from a tertiary academic referral center. J Am Acad Dermatol 2018; 78: 303–309.
- Marcoval J, Martín-Callizo C, Valentí-Medina F, Bonfill-Ortí M, Martínez-Molina L. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol 2016; 41: 741–746.
- Cohen PR, Holder WR, Tucker SB, Kono S, Kurzrock R. Sweet syndrome in patients with solid tumors. Cancer 1993; 72: 2723–2731.
- Rochael MC, Pantaleão L, Vilar EAG, Zacaron LH, Spada EQ, Xavier MHSB, et al. Sweet’s syndrome: study of 73 cases, emphasizing histopathological findings. An Bras Dermatol 2011; 86: 702–707.
- Bourke J f., Keohane S, Long C c., Kemmett D, Davies M, Zaki I, et al. Sweet’s syndrome and malignancy in the UK. Br J Dermatol 1997; 137: 609–613.
- Ghoufi L, Ortonne N, Ingen-Housz-Oro S, Barhoumi W, Begon E, Haioun C, et al. Histiocytoid Sweet syndrome is more frequently associated with myelodysplastic syndromes than the classical neutrophilic variant. Medicine (Baltimore) 2016; 95: e3033.
- Rochet NM, Chavan RN, Cappel MA, Wada DA, Gibson LE. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol 2013; 69: 557–564.
- Darrigade AS, Théophile H, Sanchez-Pena P, Milpied B, Colbert M, Pedeboscq S, et al. Sweet syndrome induced by SARS-CoV-2 Pfizer-BioNTech mRNA vaccine. Allergy 2021; 76: 3194–3196.
- Catalán-Serra I, Martín-Moraleda L, Navarro-López L, Gil-Borrás R, Pont-Sanjuán V, Ferrando-Marco J, et al. Crohn’s disease and Sweet’s syndrome: an uncommon association. Rev Espanola Enfermedades Dig Organo Of Soc Espanola Patol Dig 2010; 102: 331–337.
- Hou TY, Chang DM, Gao HW, Chen CH, Chen HC, Lai JH. Sweet’s syndrome as an initial presentation in systemic lupus erythematosus: a case report and review of the literature. Lupus 2005; 14: 399–402.
- Amouri M, Masmoudi A, Ammar M, Boudaya S, Khabir A, Boudawara T, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol 2016; 55: 1033–1039.
- Casarin Costa JR, Virgens AR, de Oliveira Mestre L, Dias NF, Samorano LP, Valente NYS, et al. Sweet syndrome: clinical features, histopathology, and associations of 83 cases. J Cutan Med Surg 2017; 21: 211–216.
- Jung EH, Park JH, Hwan Kim K, Kim JS, Sil Choi I, Byun JM, et al. Characteristics of Sweet syndrome in patients with or without malignancy. Ann Hematol 2022; 101: 1499–1508.
- Haber R, Feghali J, Gemayel ME. Risk of malignancy in histiocytoid Sweet syndrome: a systematic review and reappraisal. J Am Acad Dermatol 2020; 83: 661–663.
- Cohen PR, Talpaz M, Kurzrock R. Malignancy-associated Sweet’s syndrome: review of the world literature. J Clin Oncol Off J Am Soc Clin Oncol 1988; 6: 1887–1897.
- Kazmi SM, Pemmaraju N, Patel KP, Cohen PR, Daver N, Tran KM, et al. Characteristics of Sweet syndrome in patients with acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 2015; 15: 358–363.
- El-Khalawany M, Aboeldahab S, Mosbeh AS, Thabet A. Clinicopathologic, immunophenotyping and cytogenetic analysis of Sweet syndrome in Egyptian patients with acute myeloid leukemia. Pathol Res Pract 2017; 213: 143–153.
- Passet M, Lepelletier C, Vignon-Pennamen MD, Chasset F, Hirsch P, Battistella M, et al. Next-generation sequencing in myeloid neoplasm-associated Sweet’s syndrome demonstrates clonal relation between malignant cells and skin-infiltrating neutrophils. J Invest Dermatol 2020; 140: 1873–1876.
- Kulasekararaj AG, Kordasti S, Basu T, Salisbury JR, Mufti GJ, du Vivier AWP. Chronic relapsing remitting Sweet syndrome – a harbinger of myelodysplastic syndrome. Br J Haematol 2015; 170: 649–656.
- Merlant M, Lepelletier C, Battistella M, Vignon-Pennamen MD, Duriez P, Moguelet P, et al. Acute myeloid leukemia and myelodysplastic syndrome-associated Sweet syndrome: a comparative multicenter retrospective study of 39 patients. J Am Acad Dermatol 2021; 84: 838–840.
- Weiss EH, Ko CJ, Leung TH, Micheletti RG, Mostaghimi A, Ramachandran SM, et al. Neutrophilic dermatoses: a clinical update. Curr Dermatol Rep 2022; 11: 89–102.
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020; 383: 2628–2638.
- Zakine E, Schell B, Battistella M, Vignon-Pennamen MD, Chasset F, Mahévas T, et al. UBA1 variations in neutrophilic dermatosis skin lesions of patients with VEXAS syndrome. JAMA Dermatol 2021; 157: 1349–1354.
- Gusdorf L, Lipsker D. Neutrophilic urticarial dermatosis: an entity bridging monogenic and polygenic autoinflammatory disorders, and beyond. J Eur Acad Dermatol Venereol 2020; 34: 685–690.
- Burke N, Saikaly SK, Motaparthi K, Bender NR. Malignancy-associated Sweet syndrome presenting with simultaneous histopathologic and morphologic variants. JAAD Case Rep 2021; 14: 104–107.
- Abbas O, Kibbi AG, Rubeiz N. Sweet’s syndrome: retrospective study of clinical and histologic features of 44 cases from a tertiary care center. Int J Dermatol 2010; 49: 1244–1249.
- Neoh CY, Tan AWH, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentations and associations. Br J Dermatol 2007; 156: 480–485.