The Challenge of Basic Itch Research
Earl Carstens, Taylor Follansbee and Mirela Iodi Carstens
Department of Neurobiology, Physiology and Behavior, University of California, Davis, USA
Basic mechanisms and pathways of itch signaling are reviewed, with an emphasis on the progress to date as well as remaining challenges in translating current knowledge to the clinical treatment of chronic itch. Recent studies reveal 3 subsets of pruriceptive sensory neurons highly expressing itch-related genes. Their fibers project into the spinal cord to activate neurons expressing gastrin releasing peptide (GRP) and its receptor (GRPR), which connect to neurons that express the substance P (NK-1) receptor and project to the parabrachial nucleus and thalamus. Spinal inhibitory interneurons release GABA, glycine and dynorphin to modulate segmental itch transmission. However, nearly all pruriceptive neurons also respond to algogens such as capsaicin. Alternative theories of itch-pain discrimination, such as intensity or spatial contrast, are based on the observation that focal stimulation of nociceptive nerve endings elicits itch while more wide-spread stimulation elicits pain. These findings cloud the issue of a labeled line for itch- a long-debated but currently unresolved challenge. In higher primates there is a dichotomy of histaminergic and non-histaminergic itch-signaling pathways which is less demarcated in rodents, suggesting species differences. A cardinal symptom of chronic itch is alloknesis, i.e., mechanical or touch-evoked itch. Recent evidence indicates that low-threshold mechanosensory afferents can access the spinal itch pathway, but are normally kept in check by inhibitory interneurons expressing neuropeptide Y (NPY). In chronic itch, NPY-mediated inhibition is reduced, allowing touch to excite itch-signaling pathways. These recent advances provide novel targets for development of therapeutic strategies to relieve chronic itch.
Key words: itch; pain; labeled-line coding; gastrin releasing peptide; alloknesis.
Accepted Oct 15, 2019; Published Jan 9, 2020
Acta Derm Venereol 2020; 100: adv00023.
Corr: Prof. Earl Carstens, Department of Neurobiology, Physiology and Behavior, University of California, Davis, 1 Shields Avenue, Davis, CA 95616 USA. E-mail: eecarstens@ucdavis.edu
This paper reviews the basic mechanisms and pathways of itch signaling, emphasizing the progress to date as well as remaining challenges in translating current knowledge to the clinical treatment of chronic itch. Major questions that are addressed include: is itch signaled by a labeled-line pathway separate from that for pain; can alternative theories explain the ability to distinguish between itch and pain; are there specific markers of itch (such as gastrin releasing peptide and its receptor); are there histaminergic and non-histaminergic itch-signaling pathways? We also address challenges in understanding touch-evoked itch (alloknesis) as a symptom of chronic itch.
Like pain, acute itch provides a warning signal for the organism to scratch away insects or plant spicules from the skin surface or to dig out invasive parasites. However, chronic itch lasting > 6 weeks does not serve a useful function but instead imposes suffering, high socioeconomic costs, and reduces the quality of life. It has been estimated that itchy skin conditions such as atopic dermatitis or psoriasis affect upwards of 10% or more of the general population with associated annual health care and economic costs in the billions of dollars (1–6). Most types of chronic itch are resistant to antihistamines, so there is a pressing need to develop novel drugs and other treatment strategies. This is one of the great challenges for translational itch research. Optimism is warranted based on recent research that has led to new effective treatments for chronic itch (7).
Huge strides have been made in the past decade in our understanding of how itch is transduced and transmitted from the periphery into the central nervous system. A schematic overview of itch processing is shown in Fig. 1. A wide variety of itch mediators interact with their cognate receptors that are expressed in the free nerve endings of pruriceptive afferents in the skin. Fig. 1 provides a partial list. Histamine is the most well-known itch mediator, acting at histamine H1 and H4 receptors linked to TRPV1, the heat- and capsaicin-sensitive ion channel (8, 9), which opens to depolarize the nerve ending and thereby activate voltage sensitive sodium channels (Nav 1.7, 1.8) to initiate action potentials in the afferent fiber. Many non-histaminergic itch mediators act via TRPA1 (10), and recent reports implicate TRPV4 in histamine, serotonin and chloroquine itch transduction (11–13). Single-cell RNA sequencing has been used recently to categorize 11 subpopulations of dorsal root ganglion (DRG) cells, with 3 largely nonpeptidergic (NP) groups expressing genes associated with itch: NP1 (MrgprD), NP2 (MrgprA3), and NP3 (brain natriuretic peptide [BNP] and somatostatin) (14). MrgprD, MrgprA3, BNP and somatostatin have all been implicated in itch (15–18). In addition, neuroimmune interactions have been implicated in chronic itch. Recent studies have implicated IL-31 (19, 20), IL-4 and IL-13 (21) in itch and itch sensitization, leading to the development of biologics and antagonists that block activation of sensory neurons by cytokines (7). Clearly, improved understanding of the peripheral transduction of itch and immune function is already addressing the challenge of translating basic research into more effective treatments for chronic itch.
Pruriceptive afferent fibers transmit signals into the spinal cord dorsal horn, where they release neuropeptides including BNP (17), possibly gastrin releasing peptide (GRP) (22, 23), substance P (23), neuromedin B (24), and somatostatin (25) as well as the neurotransmitter glutamate (see below). The spinal circuitry includes excitatory interneurons that express GRP and substance P (26, 27), as well as itch-inhibitory interneurons expressing GABA, glycine and dynorphin (28–31) (Fig. 1). Projection neurons ultimately give rise to ascending itch-signaling pathways to the parabrachial nucleus and somatosensory thalamus. A high percentage of ascending projection neurons express the NK-1 receptor (32, 33). A majority of antidromically identified spinothalamic and spinoparabrachial projection neurons in rats respond to intradermal injection of pruritogens, with most also responding to the algogens capsaicin and mustard oil (34, 35). Using a double-label strategy, we observed similar proportions of retrogradely labeled spinothalamic and spinoparabrachial neurons that co-express the activity marker, c-fos, following intradermal injection of histamine, chloroquine or capsaicin (36). Finally, the spinal itch-signaling circuitry is very likely under descending modulatory influences from the brainstem, although this has only begun to be experimentally addressed (37, 38).
On the one hand, there is evidence that spinal transmission involving the neuropeptide GRP provides a specific pathway for itch transmission (discussed further below). On the other hand, based on neural recordings from peripheral and second- or higher-order neurons in the spinal cord and brain, it is evident that neurons that respond to pruritogens invariably also respond to algogens such as capsaicin, mustard oil, and other noxious stimuli. Thus, there appear to be few if any itch-specific neurons, implying that itch must be distinguished from pain (and other dysesthetic sensory qualities) by some mechanism that can decode activity in non-selective neurons. A great challenge of basic itch research is to reconcile these seemingly disparate observations to understand how itch is conveyed to the brain and how it is discriminated from pain and other sensory qualities.
There is much evidence that activation of pruritogen-sensitive primary afferent fibers elicits a sensation of itch via a specific “labeled line” pathway. An older study using electrical stimulation at discrete sites on the skin surface reported that the intensity of the evoked itch increased as a function of increasing stimulus frequency but never transitioned to pain (39). A seminal observation was that mechanically insensitive C-fibers recorded by microneurography responded to cutaneous application of histamine, such that the action potential firing pattern closely paralleled the time course of concomitant itch sensation (40). More recent studies suggest that activation of specific primary afferents elicits itch, even though the afferents respond to algogenic as well as pruritogenic stimuli. For example, MrgprA3 is a Mas-related G-protein-coupled receptor expressed in sensory nerve endings that respond to the itchy antimalarial drug chloroquine (15). When TRPV1 (the capsaicin and heat-sensitive receptor) is genetically engineered into sensory neurons expressing MrgprA3 in otherwise TRPV1-null mice, capsaicin activation of these neurons elicits itch (scratching) rather than the pain behavior that is normally elicited by capsaicin (41). Moreover, MrgprA3-expressing sensory nerves responded not only to chloroquine but also capsaicin and other chemicals, indicating that they are not itch-specific (41). Optogenetic activation of MrgprA3-expressing peripheral afferents also elicited itch-related scratching behavior (27). These findings indicate that activation of MrgprA3-expressing nerve endings elicits itch, regardless of whether they are activated by pruritic, algogenic or artificial stimuli. This implies that although the MrgprA3-expressing afferents are not exclusively activated by itchy stimuli, they access circuits at higher levels of the nervous system that selectively signal itch but not pain.
This concept is also reflected in a “population coding” theory, which is similar to the selectivity theory (Fig. 2A). Using calcium imaging of DRG sensory neurons, we found that most if not all neurons that responded to itch mediators additionally responded to capsaicin and mustard oil (42, 43). The same was true for superficial dorsal horn neurons (44). Similarly, high percentages of neurons in the ventral posterior medial and posterior triangular thalamic nuclei responded to pruritogens as well as capsaicin (45). We postulated that pruritogen- and algogen-sensitive DRG and spinal dorsal horn neurons signal itch, whereby these non-selective spinal neurons access itch-specific central mechanisms (Fig. 2A). In contrast, pruritogen-insensitive but algogen-sensitive neurons signal pain. Note that this idea still embodies separate, central labeled line mechanisms for itch and pain. Pain-evoking stimuli would activate both populations of neurons, implying that itch and pain are elicited simultaneously. In this case, pain sensation dominates due to the ability of nociceptive spinal input to activate itch-inhibitory interneurons (Fig. 2A) (46), consistent with the selectivity theory of itch. Itch perception may also be masked by stronger pain.
A role for GRP in itch was first demonstrated by a significant reduction in pruritogen-evoked scratching, but not pain behavior, in transgenic mice lacking the GRP receptor (GRPR) (22). Neurotoxic destruction of GRPR-expressing spinal neurons also significantly attenuated pruritogen-evoked scratching but not pain behavior (47). These findings were recently corroborated by the report that chemogenetic activation of GRP-expressing dorsal horn neurons elicited behavioral signs of itch but not pain (48). These data strongly support GRP and GRPR expressed in spinal neurons as itch-specific markers.
However, another recent study reported that selective activation of GRP-expressing spinal neurons elicited behavioral signs of both itch and pain (27). The authors genetically engineered TRPV1 into GRP-expressing spinal neurons in otherwise TRPV1-null mice. Intrathecal administration of capsaicin dose-dependently elicited behavioral signs of itch (scratching) as well as pain (licking). At higher doses of capsaicin (1–20 µg), pain (but not itch) responses decreased and were rescued by administration of the µ-opioid antagonist naloxone. The authors suggested that high-dose capsaicin triggered an opioid mechanism that reduced pain but not itch, and that a common population of GRP-expressing spinal neurons signals both itch and pain. This is consistent with the intensity theory of itch (Fig. 2B), which postulates that itch is signaled by a lower firing rate and pain by a higher firing rate in a common population of spinal neurons. Indeed, capsaicin elicited much higher firing rates in GRP-expressing spinal neurons compared to those elicited by the pruritogens SLIGRL, chloroquine or histamine (27). In general, capsaicin and mustard oil elicited consistently higher firing rates compared to pruritogens in spinal dorsal horn neurons, including spinothalamic projection neurons (49). Thus, there is conflicting evidence as to whether GRP- and GRPR-expressing spinal neurons are itch-specific or signal both itch and pain, a challenge to theories of itch-pain discrimination that requires future studies to resolve.
Besides GRP, neuromedin B (24), brain natriuretic peptide (BNP) (17), glutamate (50) and substance P (51) have also been implicated in the spinal transmission of itch. We found that individual intrathecal delivery of receptor antagonists of GRP (RC-3095), substance P (L-733060) or the AMPA glutamate receptor (CNQX), partially reduced scratching behavior and spinal neuronal responses to chloroquine, while a combination of all 3 antagonists completely inhibited these responses (52). This is supported by another study reporting that of dorsal horn neurons responsive to intradermal histamine or chloroquine, some responded to intrathecal delivery of BNP or GRP, but less commonly to both (53). These findings suggest that there may be parallel spinal path-ways for itch, each utilizing these neurotransmitters/neuropeptides to different extents. CNQX almost completely abolished scratching and neuronal responses to histamine, implicating glutamate as the primary spinal neurotransmitter in histaminergic itch (52). These studies provide points of intervention in the spinal cord to block the transmission of itch signals.
A further complication to our understanding of itch mechanisms is the finding that a dominant sensation of itch, together with sub-dominant nociceptive sensations (burning, stinging, pricking), were elicited by insertion of either a single histamine-loaded, or capsaicin-loaded, or native cowhage spicule into the skin (54). This implies that highly localized activation of a minimal number of nociceptive nerve endings in the skin by either pruritogenic or algogenic chemicals is sufficient to elicit a dominant sensation of itch. This supports the “spatial contrast” theory of itch, which holds that limited activation of nociceptive nerve endings is itchy, while activation of a greater number of nerve endings over a broader area (e.g., by intradermal injection of capsaicin) is painful, possibly due to disruption of a specific pattern for itch via the activation of many nociceptors. This concept was suggested as a possible mechanism of neuropathic itch following nerve injury that results in degeneration of most but not all C-fibers, such that activation of the few spared fibers elicits a sensation of itch (55). The challenge remains to explain how either itch or pain results from localized patterns of activation of nociceptive C-fibers.
It is a dogma that there are two types of itch, histaminergic and non-histaminergic. In humans, histaminergic itch is mediated by the histamine-sensitive, mechanically insensitive C-fiber afferents mentioned above (40, 56). In contrast, non-histaminergic itch can be elicited by spicules of cowhage, which contain proteases (57, 58). Cowhage excites mechanically sensitive polymodal nociceptors (56, 59). The duality of histamine- and cowhage-sensitivity applies to non-human primates as well, since intradermal injections of histamine or placement of cowhage spicules activated largely separate populations of spinothalamic tract neurons (60). However, in rodents there appears to be greater overlap in primary and secondary sensory neurons responsive to histamine and non-histaminergic itch mediators. Using calcium imaging of mouse DRG and trigeminal ganglion (TG) cells, it was variously reported that 100% (15), 50% (61) or 17–23% (62) of chloroquine-responsive cells also responded to histamine. Using in vivo recording from identified MrgprA3-expressing DRG cells in mice, 78% (7/9) responded to both histamine and chloroquine (41). Recordings from mouse spinal dorsal horn neurons revealed that 47–71% of chloroquine-responsive neurons also responded to histamine (63). In rat somatosensory thalamus, all 7 chloroquine-responsive neurons that were additionally tested with histamine responded, although it is noted that a large number of histamine-responsive thalamic neurons did not respond to chloroquine (45). These data imply that histaminergic and non-histaminergic pathways may be more segregated in humans and non-human primates compared to rodents. A challenge for the field is to understand the limitations of rodent models for translation to human itch.
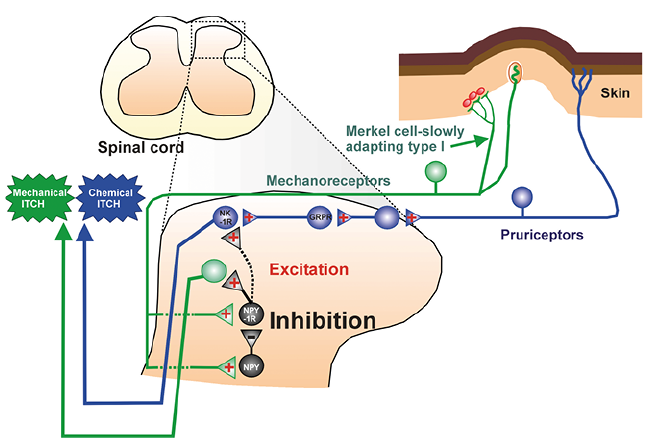
Cardinal symptoms of chronic itch are ongoing (“spontaneous”) itch, alloknesis (mechanical or touch-evoked itch), and hyperknesis (increased itch to a normally itchy or punctate mechanical stimulus). In healthy normal mice, lightly touching the skin does not elicit any behavioral signs of itch. However, following intradermal injection of histamine and other pruritogens, light touch elicits immediate scratch bouts – a model of alloknesis (64). Chemogenetic silencing of spinal neuropeptide Y (NPY) – expressing neurons led to increased touch-evoked scratching (65), and intrathecal delivery of NPY-1 receptor agonists reduced touch-evoked scratching (66), implying that the NPY-expressing neurons normally inhibit itch elicited by low-threshold mechanoreceptors. Scratching elicited by intradermal chloroquine, but not mechanical stimulation, was attenuated by antagonizing or ablating GRPR-expressing neurons, implying that mechanical itch is independent of, or converges downstream of GRP-GRPR signaling in the spinal itch circuit (Fig. 3). A reduction in the number of cutaneous Merkel cells and reduced expression of the mechanotransduction channel piezo2, as occurred in aged mice or under dry skin conditions, was associated with increased alloknesis, while chemogenetic activation of Merkel cells prevented alloknesis in dry skin (67). This suggests that Merkel cells connected to slowly adapting type I (SAI) afferents excite NPY-expressing interneurons to inhibit spinal itch transmission. It was very recently reported that activation of neurons expressing the NPY-1 receptor promotes mechanical itch (68). Mechanical itch was not affected following ablation of spinal neurons expressing the NK-1 receptor, implying that mechanical itch is transmitted via a pathway independent of that for chemical itch (Fig. 3). NPY-mediated inhibition can be overcome by mechanoreceptor activation of NPY-1 receptor-expressing neurons to drive the mechanical itch-signaling pathway under conditions in which Merkel cell-SAI input is reduced (Fig. 3).
A number of animal models have been developed to mimic various types of chronic itch and alloknesis, including atopic dermatitis, psoriasis and others (69, 70). Repeated topical application of ovalbumin induced an atopic dermatitis-like condition in mice, characterized by skin hyperplasia and lesions, increased IgG and Th2 cytokines, and importantly, increased spontaneous scratching behavior, alloknesis and hyperknesis (32). Alloknesis, but not hyperknesis or spontaneous scratching, was nearly abolished in OVA-treated mice that received intrathecal injection of substance P-saporin but not bombesin-saporin. This implies that the effect of low-threshold mechanoreceptor input to inhibit itch signaling neurons occurred downstream of GRPR-expressing neurons but requires NK-1 receptor-expressing neurons, supporting the idea that mechanoreceptive input converges onto the chemical itch-signaling pathway (Fig. 3; dashed line connecting NPY-1R to NK-1R). Consistent with this, intrathecal NPY agonists suppressed both chemically- and mechanically-evoked itch behavior (66).
Given that alloknesis is quite bothersome to patients suffering from many types of chronic itch, a challenge to the field is to better understand how low-threshold mechanosensory input interacts with spinal itch-signaling pathways and potential anti-alloknesis interventions targeting spinal NPY1 receptors.
The preceding text has identified a number of challenges arising from basic itch research to explain how itch can be discriminated from pain, and to translate our increasing knowledge of itch signaling into clinical treatment. Given the remarkable progress of the past decade and the current strong interest in itch research, several novel approaches to the treatment of chronic itch are already being used and more can be expected in the near future. Nevertheless, it has been debated for more than 100 years whether itch and pain are signaled by separate labeled-line pathways or by a common population of non-specific neurons. This debate continues unresolved up to the present, with arguments favoring both concepts.
The author’s work has been supported by a grant from the National Institute of Arthritis, Musculoskeletal and Skin Diseases, National Institutes of Health (AR057194).
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: A US population–based study. J Allergy Clin Immunol 2013; 132: 1132–1138.
View article Google Scholar - Drucker AM, Wang AR, Li WQ, Sevetson E, Block JK, Qureshi AA. The burden of atopic dermatitis: summary of a report for the National Eczema Association. J Invest Dermatol 2017; 137: 26–30.
View article Google Scholar - Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol 2017; 31: 205–212.
View article Google Scholar - Krueger GG, Bergstresser PR, Lowe NJ, Voorhees JJ, W. G. Psoriasis. J Am Acad Dermatol 1984; 11: 937–947.
View article Google Scholar - Bickers DR, Lim HW, Margolis D, Weinstock MA, Goodman C, Faulkner E, et al. The burden of skin diseases: 2004: A joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol 2006; 55: 490–500.
View article Google Scholar - Thorpe KE, Florence CS, Joski P. Which medical conditions account for the rise in health care spending? Health Aff (Millwood). Suppl Web Exclusives, W4–437–45.
View article Google Scholar - Yosipovitch G, Rosen JD, Hashimoto T. Itch: From mechanism to (novel) therapeutic approaches. J Allergy Clin Immunol 2018; 142: 1375–1390.
View article Google Scholar - Shim WS, Tak MH, Lee MH, Kim M, Kim M, Koo JY, et al. TRPV1 mediates histamine-induced itching via the activation of phospholipase A2 and 12-lipoxygenase. J Neurosci 2007; 27: 2331–2337.
View article Google Scholar - Imamachi N, Park GH, Lee H, Anderson DJ, Simon MI, Basbaum AI, et al. TRPV1-expressing primary afferents generate behavioral responses to pruritogens via multiple mechanisms. Proc Natl Acad Sci 2009; 106: 11330–11335.
View article Google Scholar - Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, et al. TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor-mediated itch. Nat Neurosci 2011; 14: 595–602.
View article Google Scholar - Akiyama T, Ivanov M, Nagamine M, Davoodi A, Carstens MI, Ikoma A, et al. Involvement of TRPV4 in serotonin-evoked scratching. J Invest Dermatol 2016; 136: 154–160.
View article Google Scholar - Chen Y, Fang Q, Wang Z, Zhang JY, MacLeod AS, Hall RP, et al. Transient receptor potential vanilloid 4 ion channel functions as a pruriceptor in epidermal keratinocytes to evoke histaminergic itch. J Biol Chem 2016; 291: 10252–10262.
View article Google Scholar - Kim S, Barry DM, Liu XY, Yin S, Munanairi A, Meng QT, et al. Facilitation of TRPV4 by TRPV1 is required for itch transmission in some sensory neuron populations. Sci Signal 2016; 9: ra71.
View article Google Scholar - Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015; 18: 145–153.
View article Google Scholar - Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell 2009; 139: 1353–1365.
View article Google Scholar - Liu Q, Sikand P, Ma C, Tang Z, Han L, Li Z, et al. Mechanisms of itch evoked by beta-alanine. J Neurosci 2012; 32: 14532–14537.
View article Google Scholar - Mishra SK, Hoon MA. The cells and circuitry for itch responses in mice. Science 2013; 340: 968–971.
View article Google Scholar - Huang J, Polgár E, Solinski HJ, Mishra SK, Tseng PY, Iwagaki N, et al. Circuit dissection of the role of somatostatin in itch and pain. Nature Neuroscience 2018; 21: 707–716.
View article Google Scholar - Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron–expressed IL-31 receptor mediates T helper cell–dependent itch: Involvement of TRPV1 and TRPA1. J Allergy Clin Immunol 2014; 133: 448–460.
View article Google Scholar - Cevikbas F, Kempkes C, Buhl T, Mess C, Buddenkotte J, Steinhoff M. Role of Interleukin-31 and Oncostatin M in Itch and Neuroimmune Communication. In: Carstens E, Akiyama T, editors. Itch: Mechanisms and Treatment. Boca Raton (FL): CRC Press; 2014, Ch. 11.
View article Google Scholar - Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell 2017; 171: 217–228.
View article Google Scholar - Sun YG, Chen ZF. A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature 2007; 448: 700–703.
View article Google Scholar - Akiyama T, Tominaga M, Davoodi A, Nagamine M, Blansit K, Horwitz A, et al. Roles for substance P and gastrin-releasing peptide as neurotransmitters released by primary afferent pruriceptors. J Neurophysiol 2013; 109: 742–748.
View article Google Scholar - Wan L, Jin H, Liu XY, Jeffry J, Barry DM, Shen KF, et al. Distinct roles of NMB and GRP in itch transmission. Sci Rep 2017; 7: 15466.
View article Google Scholar - Stantcheva KK, Iovino L, Dhandapani R, Martinez C, Castaldi L, Nocchi L, et al. A subpopulation of itch-sensing neurons marked by Ret and somatostatin expression. EMBO Rep 2016; 17: 585–600.
View article Google Scholar - Gutierrez-Mecinas M, Bell AM, Marin A, Taylor R, Boyle KA, Furuta T, et al. Preprotachykinin A is expressed by a distinct population of excitatory neurons in the mouse superficial spinal dorsal horn including cells that respond to noxious and pruritic stimuli. Pain 2017; 158: 440–456.
View article Google Scholar - Sun S, Xu Q, Guo C, Guan Y, Liu Q, Dong X. Leaky gate model: intensity-dependent coding of pain and itch in the spinal cord. Neuron 2017; 93: 840–853.
View article Google Scholar - Akiyama T, Carstens M, Carstens E. Transmitters and pathways mediating inhibition of spinal Itch-Signaling neurons by scratching and other counterstimuli. PLoS One 2011; 6: 1–10.
View article Google Scholar - Braz JM, Juarez-Salinas D, Ross SE, Basbaum AI. Transplant restoration of spinal cord inhibitory controls ameliorates neuropathic itch. J Clin Invest 2014; 124: 3612–3616.
View article Google Scholar - Kardon AP, Polgár E, Hachisuka J, Snyder LM, Cameron D, Savage S, et al. Dynorphin acts as a neuromodulator to inhibit itch in the dorsal horn of the spinal cord. Neuron 2014; 82: 573–586.
View article Google Scholar - Foster E, Wildner H, Tudeau L, Haueter S, Ralvenius WT, Jegen M, et al. Targeted ablation, silencing, and activation establish glycinergic dorsal horn neurons as key components of a spinal gate for pain and itch. Neuron 2015; 85: 1289–1304.
View article Google Scholar - Akiyama T, Nguyen T, Curtis E, Nishida K, Devireddy J, Delahanty J, et al. A central role for spinal dorsal horn neurons that express neurokinin-1 receptors in chronic itch. Pain 2015; 156: 1240–1246.
View article Google Scholar - Cameron D, Polgár E, Gutierrez-Mecinas M, Gomez-Lima M, Watanabe M, Todd AJ. The organisation of spinoparabrachial neurons in the mouse. Pain 2015; 156: 2061–2071.
View article Google Scholar - Jansen NA, Giesler GJ. Response characteristics of pruriceptive and nociceptive trigeminoparabrachial tract neurons in the rat. J Neurophysiol 2015; 113: 58–70.
View article Google Scholar - Moser HR, Giesler GJ. Characterization of pruriceptive trigeminothalamic tract neurons in rats. J Neurophysiol 2014; 111: 1574–1589.
View article Google Scholar - Akiyama T, Curtis E, Nguyen T, Carstens MI, Carstens E. Anatomical evidence of pruriceptive trigeminothalamic and trigeminoparabrachial projection neurons in mice. J Comp Neurol 2016; 524: 244–256.
View article Google Scholar - Carstens E, Iodi Carstens M, Akiyama T, Davoodi A, Nagamine M. Opposing effects of cervical spinal cold block on spinal itch and pain transmission. Itch 2018; 3: e16.
View article Google Scholar - Gao ZR, Chen WZ, Liu MZ, Chen XJ, Wan L, Zhang XY et al. Tac1-Expressing neurons in the periaqueductal gray facilitate the itch-scratching cycle via descending regulation. Neuron 2019; 101: 45–59.
View article Google Scholar - Tuckett RP. Itch evoked by electrical stimulation of the skin. J Invest Dermatol 1982; 79: 368–373.
View article Google Scholar - Schmelz M, Schmidt R, Bickel A, Handwerker HO, Torebjörk HE. Specific C-receptors for itch in human skin. J Neurosci 1997; 17: 8003–8008.
View article Google Scholar - Han L, Ma C, Liu Q, Weng HJ, Cui Y, Tang Z, et al. A subpopulation of nociceptors specifically linked to itch. Nat Neurosci 2013; 16: 174–182.
View article Google Scholar - Akiyama T, Carstens MI, Carstens E. Facial injections of pruritogens and algogens excite partly overlapping populations of primary and second-order trigeminal neurons in mice. J Neurophysiol 2010; 104: 2442–2450.
View article Google Scholar - Klein A, Carstens MI, Carstens E. Facial injections of pruritogens or algogens elicit distinct behavior responses in rats and excite overlapping populations of primary sensory and trigeminal subnucleus caudalis neurons. J Neurophysiol 2011; 106: 1078–1088.
View article Google Scholar - Akiyama T, Merrill AW, Carstens MI. Carstens E. Activation of superficial dorsal horn neurons in the mouse by a PAR-2 agonist and 5-HT: potential role in itch. J Neurosci 2009; 29: 6691–6699.
View article Google Scholar - Lipshetz B, Khasabov SG, Truong H, Netoff TI, Simone DA, Giesler GJ Jr. Responses of thalamic neurons to itch- and pain-producing stimuli in rats. J Neurophysiol 2018; 120: 1119–1134.
View article Google Scholar - Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, et al. Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron 2010; 65: 886–898.
View article Google Scholar - Sun YG, Zhao ZQ, Meng XL, Yin J, Liu XY, Chen ZF. Cellular basis of itch sensation. Science 2009; 325: 1531–1534.
View article Google Scholar - Albisetti GW, Pagani M, Platonova E, Hösli L, Johannssen HC, Fritschy JM, et al. Dorsal horn gastrin-releasing peptide expressing neurons transmit spinal itch but not pain signals. J Neurosci 2019; 39: 2238–2250.
View article Google Scholar - Davidson S, Zhang X, Khasabov SG, Moser HR, Honda CN, Simone DA, et al. Pruriceptive spinothalamic tract neurons: physiological properties and projection targets in the primate. J Neurophysiol 2012; 108: 1711–1723.
View article Google Scholar - Koga K, Chen T, Li XY, Descalzi G, Ling J, Gu J, et al. Glutamate acts as a neurotransmitter for gastrin releasing peptide-sensitive and insensitive itch-related synaptic transmission in mammalian spinal cord. Mol Pain 2011; 7: 47.
View article Google Scholar - Carstens EE, Carstens MI, Simons CT, Jinks SL. Dorsal horn neurons expressing NK-1 receptors mediate scratching in rats. Neuroreport 2010; 21: 303–308.
View article Google Scholar - Akiyama T, Tominaga M, Takamori K, Carstens MI, Carstens E. Roles of glutamate, substance P, and gastrin-releasing peptide as spinal neurotransmitters of histaminergic and nonhistaminergic itch. Pain 2014; 155: 80–92.
View article Google Scholar - Kusube F, Tominaga M, Kawasaki H, Yamakura F, Naito H, Ogawa H, et al. Electrophysiological properties of brain-natriuretic peptide- and gastrin-releasing peptide-responsive dorsal horn neurons in spinal itch transmission. Neurosci Let 2016; 627: 51–60.
View article Google Scholar - LaMotte RH, Shimada SG, Green BG, Zelterman D. Pruritic and nociceptive sensations and dysesthesias from a spicule of cowhage. J Neurophysiol 2009; 101: 1430–1443.
View article Google Scholar - Steinhoff M, Oaklander AL, Szabó IL, Ständer S, Schmelz M. Neuropathic itch. Pain 2019; 160: S11–S16.
View article Google Scholar - Namer B, Carr R, Johanek LM, Schmelz M, Handwerker HO, Ringkamp M. Separate peripheral pathways for pruritus in man. J Neurophysiol 2008; 100: 2062–2069.
View article Google Scholar - Shelley WB, Arthur RP. Mucunain, the active pruritogenic proteinase of cowhage. Science 1955; 122: 469–470.
View article Google Scholar - Reddy VB, Iuga AO, Shimada SG, LaMotte RH, Lerner EA. Cowhage-evoked itch is mediated by a novel cysteine protease: a ligand of protease-activated receptors. J Neurosci 2008; 28: 4331–4335.
View article Google Scholar - Johanek LM, Meyer RA, Friedman RM, Greenquist KW, Shim B, Borzan J, et al. A role for polymodal C-fiber afferents in nonhistaminergic itch. J Neurosci 2008; 28: 7659–7669.
View article Google Scholar - Davidson S, Zhang X, Yoon CH, Khasabov SG, Simone DA, Giesler GJ Jr. The itch-producing agents histamine and cowhage activate separate populations of primate spinothalamic tract neurons. J Neurosci 2007; 27: 10007–10014.
View article Google Scholar - Akiyama T, Tominaga M, Davoodi A, Nagamine M, Blansit K, Horwitz A, et al. Cross-sensitization of histamine-independent itch in mouse primary sensory neurons. Neuroscience 2012; 226: 305–312.
View article Google Scholar - Roberson DP, Gudes S, Sprague JM, Patoski HA, Robson VK, Blasl F, et al. Activity-dependent silencing reveals functionally distinct itch-generating sensory neurons. Nat Neurosci 2013; 16: 910–918.
View article Google Scholar - Akiyama T, Tominaga M, Takamori K, Carstens MI, Carstens E. Role of spinal bombesin-responsive neurons in nonhistaminergic itch. J Neurophysiol 2014; 112: 2283–2289.
View article Google Scholar - Akiyama T, Carstens MI, Ikoma A, Cevikbas F, Steinhoff M, Carstens E. Mouse model of touch-evoked itch (alloknesis). J Invest Dermatol 2012; 132: 1886–1891.
View article Google Scholar - Bourane S, Duan B, Koch SC, Dalet A, Britz O, Garcia-Campmany L, et al. Gate control of mechanical itch by a subpopulation of spinal cord interneurons. Science 2015; 350: 550–554.
View article Google Scholar - Gao T, Ma H, Xu B, Bergman J, Larhammar D, Lagerström MC. The Neuropeptide Y System Regulates Both Mechanical and Histaminergic Itch. J Invest Dermatol 2018; 138: 2405–2411.
View article Google Scholar - Feng J, Luo J, Yang P, Du J, Kim BS, Hu H. Piezo2 channel-Merkel cell signaling modulates the conversion of touch to itch. Science 2018; 360: 530–533.
View article Google Scholar - Acton D, Ren X, Di Costanzo S, Dalet A, Bourane S, Bertocchi I, et al. Spinal neuropeptide Y1 receptor-expressing neurons form an essential excitatory pathway for mechanical itch. Cell Rep 2019; 28: 625–639.
View article Google Scholar - Bautista DM, Wilson SR, Hoon MA. Why we scratch an itch: the molecules, cells and circuits of itch. Nat Neurosci 2014; 17: 175–182.
View article Google Scholar - Kim D, Kobayashi T, Nagao K. Research techniques made simple: mouse models of atopic dermatitis. J Invest Dermatol 2019; 139: 984–990.
View article Google Scholar