1Department of Dermatology, Peking University Shenzhen Hospital, Shenzhen Key Laboratory for Translational Medicine of Dermatology, Shenzhen 518036, and 2Department of Dermatology, Peking University First Hospital, Beijing Key Laboratory of Molecular Diagnosis on Dermatoses, National Clinical Research Center for Skin and Immune Diseases, Beijing 100034, China.*E-mails: zhimiaolin@bjmu.edu.cn; yubomd@163.com
#These authors contributed equally.
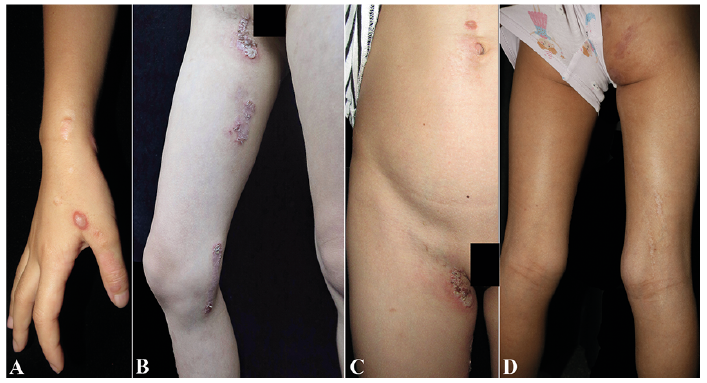
An 8-year-old Chinese girl had a 7-year history of blisters and erythema affecting the right side of her trunk and extremities (Fig. 1). These itchy lesions first appeared on the dorsum of her right hand at the age of 1 year, and they easily ruptured, leading to macerated or crusted erosions/plaques. Hyper- or hypopigmentation was noted after the lesions healed. The lesions progressed to involve her right body generally, with a linear distribution on her right extremities. The lesions exacerbated in summer. No extracutaneous abnormalities were found. Her father and grandfather had mild and recurrent blisters and erosions distributed symmetrically at intertriginous sites, with an onset age of around 30 years. A biopsy specimen was obtained from the lesion on the abdomen.
What is your diagnosis? See the next page for the answer.
Acta Derm Venereol 2020; 100: adv000115.
Diagnosis: Type 2 segmental Hailey-Hailey disease
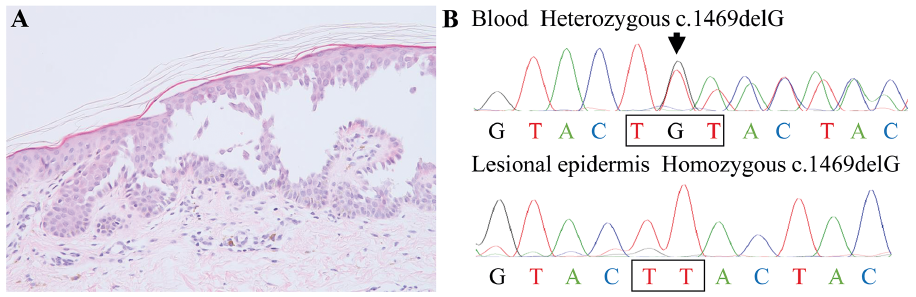
Histopathological examination revealed a suprabasal cleft with acantholysis, showing the sign of a ‘dilapidated brick wall’, as well as focal perivascular infiltration of lymphocytes in the superficial dermis (Fig. 2A). Genetic testing using DNA from the peripheral blood of the proband and her father revealed a heterozygous frameshift mutation c.1469delG (p.C490Lfs*11) in ATP2C1 (Fig. 2B). Further, Sanger sequencing using DNA extracted from the keratinocytes of the ‘dilapidated brick wall’ obtained by laser-captured microdissection revealed only a mutant allele (c.1469delG) (Fig. 2B), whereas in normal skin, equivalent peaks of the mutant and wild-type alleles were detected, as seen in the sequencing of blood DNA. Thus, a diagnosis of type 2 segmental Hailey-Hailey disease (HHD) was confirmed.
HHD (OMIM 169600) is a rare autosomal dominant genodermatosis caused by mutations in ATP2C1 (OMIM 604384), a gene coding the Golgi-associated Ca2+/Mn2+ ATPase hSPCA1 (1). There are two types of segmental HHD. Type 1 segmental HHD involves a de novo postzygotic somatic mutation that leads to heterozygosity of ATP2C1 in the segmentally distributed lesions, whereas type 2 segmental HHD, which is extremely rare, is caused by a germline mutation in combination with somatic loss of heterozygosity (LOH) in ATP2C1 during early embryogenesis (2). Therefore, type 2 segmental HHD is characterised by early onset, usual occurrence in infancy, and more pronounced lesions. Before the onset of canonical lesions of HHD, the lesions of type 2 segmental HHD display a unilateral, usually linear distribution of blisters. Since the first description by Vakilzadeh et al. (3) in 1985 and the reclassification by König et al. (4) in 2000, only 7 cases of type 2 segmental HHD have been reported, including 2 cases of relapsing linear acantholytic dermatosis (1, 2, 5). A review of these cases suggested that type 2 segmental HHD could also affect noncanonical sites, including the trunk, extremities, scalp, and anogenital and supraorbital regions (1, 2, 4, 5). This was also noted in our proband who presented with generalised distribution of lesions involving noncanonical sites, such as the wrist and dorsal hand, as well as the antecubital and popliteal fossae (6, 7). This unusual distribution pattern obscures an accurate diagnosis.
In 2004, Poblete-Gutiérrez et al. (1) first reported the molecular mechanisms of type 2 segmental HHD. Specifically, LOH due to postzygotic mitotic recombination caused additional loss of the paternal wild-type ATP2C1 allele, leading to the unilateral distribution of superimposed linear lesions in the proband (1). Subsequently, the diagnosis of type 2 segmental involvement was mainly based on clinical presentation, including in a recently reported case with systematised bilateral arrangement (2, 5). Here, we present the second case of type 2 segmental HHD with genetic evidence. We found that the loss of both ATP2C1 alleles in our proband could be associated with a much more pronounced and early-onset phenotype, which is supported by a lethal phenotype in homozygous knock-out of ATP2C1 in mice (8). However, further studies are needed to elucidate the exact mechanism, e.g., whole-exome or whole-genome sequencing to elucidate whether the LOH in the present patient was caused by somatic homologous recombination or a second hit mutation, such as a large deletion including the region of wild-type ATP2C1.
The lesions in the proband resolved with topical glucocorticoids within 2 months. However, long-term follow-up is required to determine whether lesions will relapse in summer and whether a great relief of lesions will happen in her 20s as previously reported (1, 4).
We thank the patient for granting permission to publish this information. This work was supported by the National Natural Science Foundation of China (grant nos. 81573032 and 81872515), Fok Ying Tung Education Foundation (grant no. 161038), and the fund of the ‘San?ming’ Project of Medicine in Shenzhen (no. SZSM201812059).
The authors have no conflicts of interest to declare.