Mycosis fungoides (MF) is the most common primary cutaneous T-cell lymphoma (CTCL), typically originating from CD4+, and less commonly, from CD8+ T-cells. Several forms or subtypes of MF have been described, including rare ones, such as poikilodermatous MF (PMF). PMF is currently considered a rare clinicopathological variant of MF, formerly known as poikiloderma vasculare atrophicans or parapsoriasis variegate (1). Clinically, it is typically characterized by large patches or plaques with areas of hyperpigmentation or hypopigmentation, atrophy and telangiectasias (2). These patches or plaques may be asymptomatic, mildly pruritic, or be accompanied by a stinging sensation. Of note, poikilodermatous lesions frequently coexist with classic MF lesions. PMF lesions may sometimes be confusing and raise differential diagnosis with several inflammatory and non-inflammatory conditions of the skin, including lichen ruber planus, and morphea, rendering clinicopathological correlation or even multiple biopsies essential (3).
Treatment recommendations for PMF are similar to those for classic MF, with psoralen with ultraviolet A (PUVA) being the most common first-line therapy. In addition, previous studies have shown that PMF has an overall favourable prognosis and a low risk of disease progression (2, 3). We report here 2 cases of PMF with a chronic course and poor response to treatment, focusing on the diagnostic and treatment challenges.
CASE REPORTS
Case 1. A 52-year-old woman presented with patches consisting of mottled hypopigmentation and hyperpigmentation with atrophy and telangiectasias on her face, upper and lower extremities, and trunk (Fig. 1A). The lesions had first presented 10 years earlier. The patient was treated as pigmented lichen planus with short courses of systemic and topical steroids, with no response. A skin biopsy was performed, and histological examination revealed mildly atrophic epidermis and a lichenoid lymphocytic infiltration of the papillary dermis, accompanied by epidermotropism (Fig. 1B). Atypical lymphoid cells presented a clear halo. Telangiectatic vessels and pigment incontinence were evident. Immunohistochemical examination revealed atypical lymphoid cells of T cell origin, positive to CD3 and predominantly to CD8 (Fig. 1C) and negative to CD5 and CD7 antibodies. Interestingly, there were a few FOXP3+ cells, many of which presented atypia (Fig. 1D). Periodic acid–Schiff (PAS) and Gram histochemical stains did not reveal any microorganisms. Based on the clinical and histopathological findings a diagnosis of PMF was made. The patient was initially treated with interferon-α and then with bexarotene, with no improvement. A second skin biopsy was performed, showing similar morphological and immunohistochemical findings. In addition, T-cell receptor (TCR)-β gene rearrangement analysis revealed a monoclonal TCR rearrangement. The patient underwent total skin electron beam therapy (TSEB), and currently remains stable on a low dose of bexarotene.
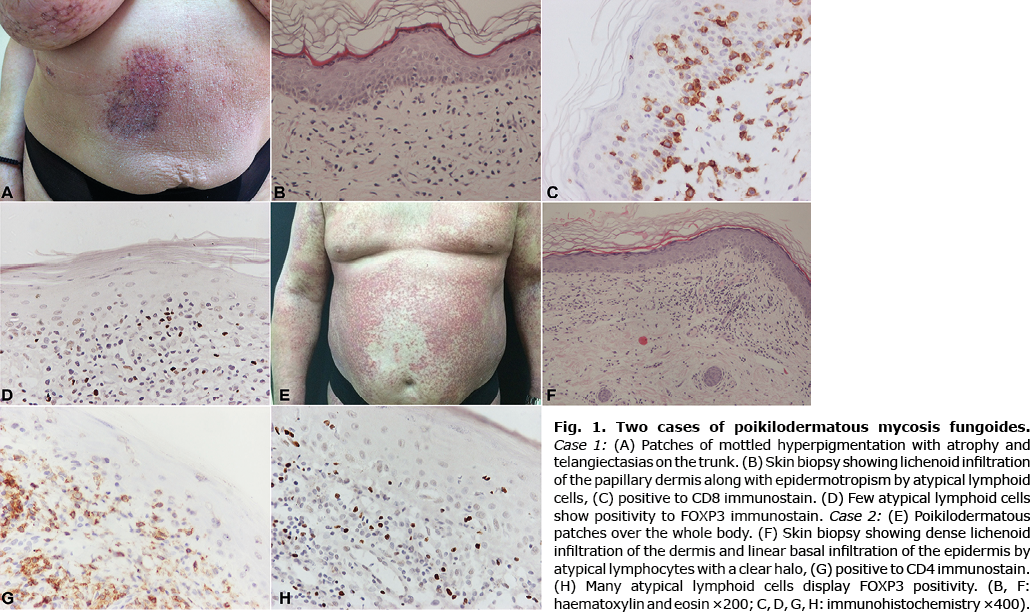
Case 2. 62-year-old man presented with poikilodermatous patches all over his body (Fig. 1E), leaving his face unaffected. The onset of lesions was reportedly 6 years earlier. Biopsy of the lesions revealed a mildly atrophic epidermis and a band-like lymphocytic infiltration of the papillary dermis. Few lymphocytes presented a halo and epidermotropism. Sparse necrotic keratinocytes were noted. Immunohistochemical evaluation revealed CD3 and CD4 positivity of the intraepidermal lymphoid cells. There was no deletion of CD5, CD7, or CD2 T cell markers. The histological findings were consistent with PMF. Clinicopathological correlation and close follow-up of the patient were suggested. The initial treatment plan, involving cyclosporine, and intravenous prednisone pulses, failed. A second biopsy, almost 5 years later, showed mildly atrophic epidermis and a lichenoid lymphocytic infiltration. Lymphocytes showed atypia and a clear halo. Epidermotropism and linear basement lymphocytic infiltration were prominent (Fig. 1F). Immunohistochemical analysis revealed CD3 and CD4 immunoreactivity (Fig. 1G) and negativity to CD8 antibody. In addition, many atypical cells displayed FOXP3 positivity (Fig. 1H). PAS and Gram histochemical stains did not reveal any microorganism. Molecular analysis of both tissue specimens showed oligoclonality. The overall findings were consistent with PMF. The patient was treated with PUVA, acitretin, methotrexate, interferon-α, combined with low isotretinoin and acitretin, and, currently, with methotrexate again, with no clinical improvement, but also no disease progression or stage evolution.
DISCUSSION
Two cases with similar clinical and histological findings are presented. The histological and molecular findings were more typical of MF in the first case. Comparison of the cases reveals a morphologically analogous atypical lymphocytic population with peripheral clear halo, demonstrating epidermotropism, lichenoid cutaneous infiltration, albeit immunophenotypic and molecular diversity. However, it is well known that MF demonstrates significant clinical and histological variation. In this context, under the term PMF, there have been reported cases with similar clinical, but different immunophenotypic, findings (4). The most commonly reported immunophenotype in PMF is predominantly CD4+, although predominantly CD8+ or double-negative (CD4–/CD8–) cases have also been described (2, 4). Furthermore, although molecular analysis of clonal TCR gene rearrangements is considered a complementary diagnostic tool for cutaneous lymphomas, it is well-known that detection of monoclonality of a T-cell infiltrate is absent in many cases of true lymphomas and present in a considerable number of pseudolymphomas (3, 5, 6).
FOXP3 positivity has been investigated mainly on the basis of the prognostic role of Tregs in the CTCL microenvironment, including that of MF (7, 8). Specifically, an increased number of FOXP3+ Tregs has been found in early patch/plaque as opposed to late tumour MF stage, and has been associated with a favourable prognosis (9). CTCL neoplastic cells do not typically express FOXP3 protein. This could be explained by the well-known antiproliferative effect of the FOXP3 protein, which would go against the proliferative nature of CTCL neoplastic cells. Paradoxically, FOXP3 positivity in a proportion of atypical intraepidermal lymphoid cells has been previously described in different stages of classic MF (8, 10). Furthermore, it has been shown that neoplastic T cells in Sézary syndrome primarily express the low molecular weight isoforms of FOXP3 (11). Moreover, according to a recent study, C-terminal cleaved forms of FOXP3 exert no antiproliferative effect, thus potentially settling the aforementioned paradox (12). Interestingly, another study demonstrated that Staphylococcus aureus enterotoxins can induce FOXP3 expression in neoplastic T cells in Sézary syndrome (13). This could indicate that the variable frequency of FOXP3 expression by neoplastic T cells might reflect differences in colonization or infection of the patient’s skin by enterotoxin-producing S. aureus.
To our knowledge, this is the first case reporting FOXP3 expression by atypical lymphoid cells in PMF. It is unknown if this is a consistent characteristic of PMF with potential diagnostic or prognostic value. FOXP3+ atypical cells were present in both biopsies of each patient, raising questions about their origin and possible implication in the resistance to treatment, which needs further investigation. In addition, since immunohistochemistry cannot discriminate between the wild-type and the molecular isoforms of FOXP3, it might be worth exploring whether it is the smaller, non-suppressive isoform of the protein that is expressed by the atypical lymphoid cells in PMF.
Similarly to classic MF, the choice of treatment for PMF depends on the stage of the disease and includes skin-directed therapies, such as topical corticosteroids, bexarotene, carmustine, mechlorethamine, PUVA, and radiation, which can be used in combination with systemic therapies, such as retinoids or interferon-alpha (2, 3). Notably, in PMF a high proportion of patients present many years after the onset of lesions (1, 14). The older the lesions are, the more resistant to treatment is the depigmentation. Consequently, as the overall prognosis is favourable, many patients stop treatment.
In conclusion, PMF remains a challenging clinical type of MF to diagnose and treat. Further data on the expression of FOXP3 in PMF lesions may clarify the potential diagnostic role of this marker, as well as its possible association with treatment resistance.
The authors have no conflicts of interest to declare.
REFERENCES
- Pankratov O, Gradova S, Tarasevich S, Pankratov V. Poikilodermatous mycosis fungoides: Clinical and histopathological analysis of a case and literature review. Acta Dermatovenerol Alp Pannonica Adriat 2015; 24: 37–41.
- Abbott RA, Sahni D, Robson A, Agar N, Whittaker S, Scarisbrick JJ. Poikilodermatous mycosis fungoides: A study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol 2011; 65: 313–319.
- Jerković Gulin S, Čeović R, Ilić I, Bradamante M, Mokos ZB, Kostović K. Poikilodermatous mycosis fungoides – rare entity, different treatment modalities. Acta Dermatovenerologica Croat 2018; 26: 48–52.
- Berg RV, Valente NYS, Fanelli C, Wu I, Pereira J, Zatz R, et al. Poikilodermatous mycosis fungoides: comparative study of clinical, histopathological and immunohistochemical features. Dermatology 2020; 236: 117–122.
- Mitteldorf C, Kempf W. Cutaneous pseudolymphoma – a review on the spectrum and a proposal for a new classification. J Cutan Pathol 2020; 47: 76–97.
- van Krieken JHJM, Langerak AW, Macintyre EA, Kneba M, Hodges E, Sanz RG, et al. Improved reliability of lymphoma diagnostics via PCR-based clonality testing: - Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia 2007; 21: 201–206.
- Klemke CD, Fritzsching B, Franz B, Kleinmann EV, Oberle N, Poenitz N, et al. Paucity of FOXP3+ cells in skin and peripheral blood distinguishes Sézary syndrome from other cutaneous T-cell lymphomas. Leukemia 2006; 20: 1123–1129.
- Fried I, Cerroni L. FOXP3 in sequential biopsies of progressive mycosis fungoides. Am J Dermatopathol 2012; 34: 263–265.
- Gjerdrum LM, Woetmann A, Odum N, Burton CM, Rossen K, Skovgaard GL, et al. FOXP3+ regulatory T cells in cutaneous T-cell lymphomas: association with disease stage and survival. Leukemia 2007; 21: 2512–2518.
- Berger CL, Tigelaar R, Cohen J, Mariwalla K, Trinh J, Wang N, et al. Cutaneous T-cell lymphoma: malignant proliferation of T-regulatory cells. Blood 2005; 105: 1640–1647.
- Krejsgaard T, Gjerdrum LM, Ralfkiaer E, Lauenborg B, Eriksen KW, Mathiesen AM, et al. Malignant Tregs express low molecular splice forms of FOXP3 in Sézary syndrome. Leukemia 2008; 22: 2230–2239.
- Elhage R, Cheraï M, Levacher B, Darrasse-Jeze G, Baillou C, Zhao X, et al. C-terminal cleavage of human Foxp3 at a proprotein convertase motif abrogates its suppressive function. Scand J Immunol 2015; 81: 229–239.
- Willerslev-Olsen A, Buus TB, Nastasi C, Blümel E, Gluud M, Bonefeld CM, et al. Staphylococcus aureus enterotoxins induce FOXP3 in neoplastic T cells in Sézary syndrome. Blood Cancer J 2020; 10: 57.
- Liu C, Tan C. 34 years duration of poikilodermatous lesion. Postep Dermatologii i Alergol 2020; 37: 438–440.