1Department of Dermatology and Allergology, Philipp University, Baldingerstraße, DE-35043 Marburg, 2Institute of Human Genetics, and 3Department of Dermatology, Medical Center – University of Freiburg, Freiburg, Germany. E-mail: juratlih@med.uni-marburg.de
#These authors contributed equally.
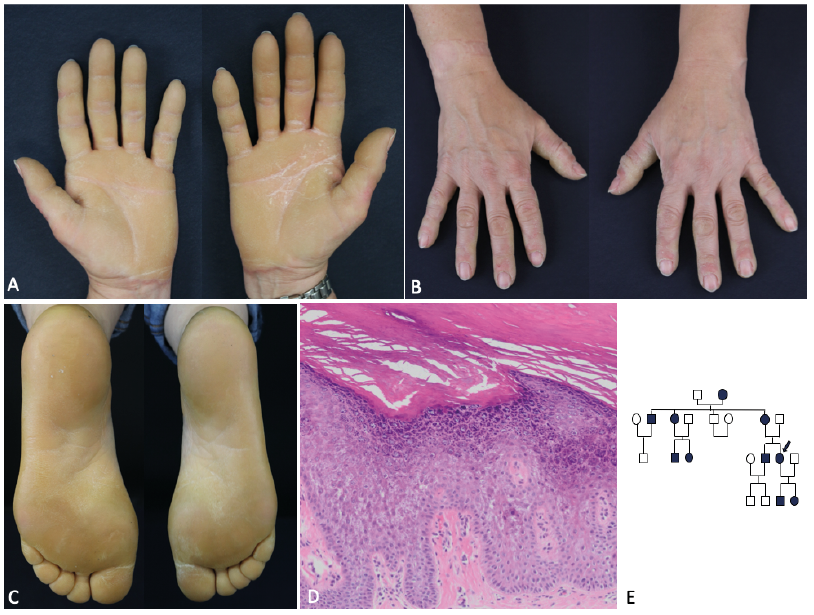
An otherwise healthy 49-year-old Caucasian woman presented to our dermatology department with bilateral diffuse hyperkeratosis on her palms and soles with a sharp cut-off and erythematous border (Fig. 1A–C). She did not report any pain or pruritus. The lesions started in infancy, and the patient reported similar skin lesions in her grandmother, mother, aunt, uncle, brother, daughter and son (Fig. 1E). A complete physical examination revealed otherwise unremarkable findings, and basic laboratory test results were within normal limits. She was not taking any medications. Histopathological examination of a punch biopsy from the palmar skin showed orthokeratotic hyperkeratosis and acanthosis. Vacuolar changes were identified in the granular cell layer, which is thickened. In the papillary dermis a mild perivascular infiltrate was observed (Fig. 1D). Following written informed consent for genetic analysis, peripheral blood samples were obtained from the proband and her daughter and DNA was extracted using standard procedures and next generation sequencing (NGS) multi-gene-panel sequencing was performed.
What is your diagnosis? See next page for answer.
Acta Derm Venereol 2020; 100: XX–XX.
Diagnosis: Epidermolytic palmoplantar keratoderma of Unna-Thost-Vörner associated with a new mutation in the keratin 1 gene: MIM #144200
Diffuse epidermolytic (keratinopathic) palmoplantar keratosis (PPK of Unna-Thost-Vörner) is the most frequently occurring form of PPK, being caused by a heterozygous mutations in one of the keratin genes KRT1 or KRT9 (1, 2). PPK becomes apparent at birth or early childhood (3). Clinically it is characterized by marked hyperkeratosis on the surface of soles and palms and, histopathologically, it is identified by hyperkeratosis and vacuolar degeneration of cells in the granular and spinous layers of the epidermis. These alterations are due to the physical cell damage by weakness of the cytoskeleton caused by disrupted formation of keratin intermediate filaments (4).
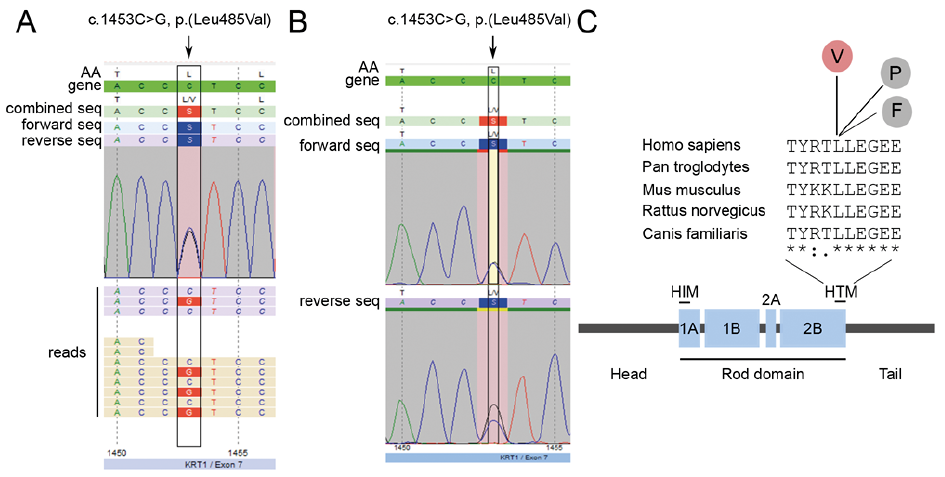
NGS analyses revealed the heterozygous missense variant c.1453C>G, p.(Leu485Val) in exon 7 of KRT1 in the index patient (Fig. 2, inset A) and the affected daughter also carried this variant (Fig. 2, inset B). The amino acid leucine at position 485 is part of a highly conserved helix termination motif (HTM) at the end of the 2B domain of the KRT1 protein (Fig. 2, inset C). To date, at position 485, 2 pathogenic changes of the amino acid leucine have been described. Arin et al. identified a substitution of leucine by proline in a patient with severe generalized keratinopathic ichthyosis that includes palmoplantar involvement (5). By contrast, a patient with the mutation c.1453C>T, p.(Leu485Phe) (6) as well as the current patient with the mutation c.1453C>G, p.(Leu485Val) presented with diffuse PPK only. In all 3 cases the neutral, non-polar amino acid leucine is replaced by another neutral, non-polar amino acid. Presumably the substitution by proline results in a more severe phenotype than that of valine and phenylalanine due to the property of proline to form kinks in α-helices, thereby influencing protein secondary structure (7). Moreover, at other amino acid positions different substitutions in the KRT1 protein, resulting in a severe or mild phenotype, e g. p.(Glu478Lys) (8) and p.(Glu478Asp) have been described (9). Remarkably, the same mutation can lead to different phenotypes in different patients (5, 10, 11). This suggests that other genetic factors might have an impact on the patients’ phenotypes.
Patients with diffuse PPK usually benefit from daily to weekly bath soaks, followed by gentle mechanical scale removal with pumice stone and creams chosen according to patient preference.