SHORT COMMUNICATION
Hereditary Angioedema Type 1 and 2 in Finland: Incidence, Prevalence, and Preceding Diagnoses
Andreas SANDBERG1, Mariann LASSENIUS2, Ville VIHERVAARA1, Iiro TOPPILA2 and Laura HUILAJA3
1Takeda Oy, Ilmalantori 1, P.O. Box 1406, FIN-00101 Helsinki, 2Medaffcon Oy, Espoo, and 3Department of Dermatology, Oulu University Hospital and Research Unit of Clinical Medicine, University of Oulu, Oulu, Finland. E-mail: andreas.sandberg@takeda.com
Citation: Acta Derm Venereol 2024; 104: adv24176. DOI: https://doi.org/10.2340/actadv.v104.24176.
Copyright: © 2024 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/)
Submitted: Oct 23, 2023. Accepted after review: Mar 4, 2024. Published: May 6, 2024
INTRODUCTION
Hereditary angioedema (HAE) is a rare genetic autosomal dominant disorder that manifests with episodes of cutaneous or submucosal oedema, most commonly affecting the skin, abdomen, and upper respiratory tract, occurring spontaneously or in response to minimal triggers (1, 2). Some of these episodes can be fatal if left untreated. Up to 50% of HAE patients experience at least one episode of potentially life-threatening, asphyxiating laryngeal oedema (3). HAE is often a result of C1-esterase inhibitor (C1-INH) deficiency, affecting the contact activation and complement systems and causing leakage of fluids from the circulation to tissues (4). Most HAE patients present with low C1-INH levels (HAE type 1, ≈85% of cases) or dysfunctional protein (HAE type 2, ≈15% of cases). In a small subset of HAE patients, the levels and function of C1-INH are normal (HAE-nC1-INH, < 1% of cases). Globally the prevalence of HAE type 1 and 2 (HAE-1/2) has been estimated to be between 1:50,000 and 1:100,000 but the true prevalence is unknown (5). Ethnicity does not seem to influence the prevalence and HAE has been observed all around the world (5). HAE is often poorly recognised due to the rareness and nonspecific symptoms causing delays in diagnosis. Even if the time from symptom onset to diagnosis has been improving, the diagnosis time in patients born between 1980 and 1990 was still over 1.4 years for half of the patients (6).
We investigated the incidence and prevalence of HAE-1/2 in Finland, which has not been described before. Moreover, diagnoses of patients 5 years prior to HAE diagnosis were investigated to elucidate possible symptoms relevant for patient identification.
METHODS
Patients with at least 2 recorded diagnoses for HAE-1/2 (D84.11, D84.12) were searched from the national secondary care register for health care maintained by the Finnish Institute for Health and Welfare. Incident patients were included from first diagnosis after 1 January 2010, and all recorded diagnoses preceding the first HAE diagnosis were evaluated 5 years a priori. For prevalence, patients with an HAE diagnosis after 2005 until end of 2021 or death were included. Both incidence and prevalence were reported annually per 100,000 persons.
RESULTS
In Finland, 103 new patients were diagnosed with HAE-1/2 within the 10-year study period. The mean incidence of HAE-1/2 was 0.16/100,000, showing an annual variability between 0.07 and 0.22/100,000 (4 to 12 new cases annually), Fig. 1. The prevalence of HAE-1/2 patients increased over the follow-up time from 0.8 to 2.6/100,000 persons, equalling 144 patients at the end of 2021.
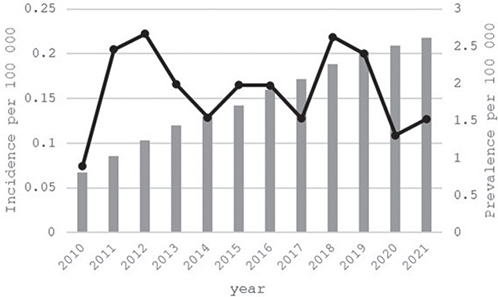
Fig. 1. Annual incidence (black line) and prevalence (grey bar) of HAE-1/2 in 2010–2021 in Finland per 100,000 persons.
The patient characteristics at diagnosis are described in Table I. Mean age at the time of diagnosis was 39 years, with 22.5% diagnosed at ≤18 years, and 13.6% before or at the age of 12. Prior to the first HAE diagnosis, 37% had a diagnosis of “adverse effects, not elsewhere classified” (incl. angioedema), followed by “exposure to unspecific factor” in 21%, “abdominal and pelvic pain” in 21%, diagnosis of “special examinations” in 18%, and “unspecific oedema” in 17% of patients (Table I).
| N | 103 |
| Age at diagnosis, mean (SD) | 38.6 (23.0) |
| Under 12 years at diagnosis, n (%) | 14 (13.6) |
| Age 12–18 years at diagnosis, n (%) | 9 (8.7) |
| Females, n (%) | 61 (59.2) |
| Diagnoses 5 years prior to HAE-1/2 diagnosis, n (%) | |
| Adverse effects, not elsewhere classified incl. angioedema (T78)* | 38 (37.0) |
| Exposure to unspecified factor (X59)* | 21 (20.4) |
| Abdominal and pelvic pain (R10)* | 21 (20.4) |
| Other special examinations and investigations of persons without complaint or reported diagnosis (Z01)* | 19 (18.5) |
| Oedema, not elsewhere classified (R60)* | 18 (17.5) |
| Other immunodeficiencies (D84)* | 16 (15.5) |
| Medical observation and evaluation for suspected diseases and conditions (Z03)* | 12 (11.7) |
| Urticaria (L50)* | 6 (5.8) |
| Asthma (J45)* | 6 (5.8) |
| Dorsalgia (M54)* | 3 (2.9) |
| *The International Classification of Diseases (ICD)-10 codes in brackets | |
DISCUSSION
Our study investigated for the first time the incidence and prevalence of HAE-1/2 patients in Finland. The diagnosed HAE incidence has not been described accurately in modern publications previously. In our study the incidence seemed quite stable, especially considering that HAE is a rare disease, and a few new patients (e.g. a new HAE family) have a big impact on the calculated annual incidence in a small country like Finland. Prevalence, however, increased notably over time. The prevalence of 2.6 per 100,000 at the end of 2021 in Finland is higher than the 1.5 per 100,000 that has been observed in the other Nordic countries including Sweden, Denmark, and Norway (7–10), and the prevalence observed in other European countries including Italy (1.51 per 100,000) and Greece (1.07 per 100,000) (7). Founder effect and past genetic isolation of Finnish people may explain the higher prevalence we observed, but also differences in study designs as well as the years covered by different studies, as patient identification (including genetic testing) has been improving over time (6). The results are nevertheless in line with the higher end of the global prevalence estimates of 1:50,000–1:100,000 (5).
The rise in yearly prevalence is likely a combination of stable incidence and decreased mortality, e.g. due to more effective HAE treatment options (1). Increasing disease awareness and diagnostic testing may also be reflected in the increased prevalence. Before the first HAE diagnosis, patients most commonly had unspecific diagnoses related to oedema and pain, which may be associated with an oedemic response to minimal external triggers. Of note, 37% had a diagnosis of angioedema before the first HAE diagnosis, 17% a diagnosis of oedema, whereas 6% had been diagnosed with urticaria, suggesting misdiagnosed HAE or unrelated urticaria. The diagnoses preceding the first HAE diagnosis highlight the diffuse variety of symptoms related to HAE, and they differ substantially from what typically has been seen in dermatological patients with similar age distribution; only asthma was shared as a co-diagnosis between our study and studies evaluating comorbidities in alopecia areata and hyperhidrosis (11, 12). Even if direct comparisons cannot be made, the list of prior diagnoses poses a starting point for possible patient identification that can be confirmed by laboratory tests.
In our registry study we could not verify the correctness of the recorded diagnoses, but by excluding patients with only one HAE diagnosis we were able to minimize the risk of including patients with erroneous HAE diagnosis. Overall, Finnish healthcare registries are highly regarded and the accuracy of dermatological diagnoses in hospital registries has been shown in several studies (13–15).
To conclude, HAE-1/2 is a rare condition, with an increasing prevalence, and clinically special focus should be given to patients with oedema, especially if the causative factor remains elusive, to determine possible HAE diagnosis.
ACKNOWLEDGEMENT
Conflicts of interest and funding: AS and VV are employees of Takeda Oy, which funded this research. ML and IT are employees of Medaffcon Oy, which has received funding for the conduct of the study. LH has received educational grants from Takeda, Janssen-Cilag, Novartis, AbbVie, and LEO Pharma, honoraria from Lilly, Sanofi, Novartis, Abbvie, Leo Pharma, Boehringer Ingelheim, Takeda, BioCryst, CSL Behring, and Orion Pharma for consulting and/or speaking, and is an investigator for Abbvie and Amgen.
REFERENCES
- Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol 2012; 130: 692–697.
- Busse PJ, Christiansen SC, Riedl MA, Banerji A, Bernstein JA, Castaldo AJ, et al. US HAEA Medical Advisory Board 2020 guidelines for the management of hereditary angioedema. J Allergy Clin Immunol Pract 2021; 9: 132–150.e3.
- Bork K, Hardt J, Schicketanz KH, Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med 2003; 163: 1229–1235.
- Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema: the 2021 revision and update. Allergy 2022; 77: 1961–1990.
- Lumry WR, Settipane RA. Hereditary angioedema: epidemiology and burden of disease. Allergy Asthma Proc 2020; 41: S08–S13.
- Zanichelli A, Magerl M, Longhurst HJ, Aberer W, Caballero T, Bouillet L, et al. Improvement in diagnostic delays over time in patients with hereditary angioedema: findings from the Icatibant Outcome Survey. Clin Transl Allergy 2018; 8: 42.
- Aygören-Pürsün E, Magerl M, Maetzel A, Maurer M. Epidemiology of bradykinin-mediated angioedema: a systematic investigation of epidemiological studies. Orphanet J Rare Dis 2018; 13: 73.
- Bygum A. Hereditary angio-oedema in Denmark: a nationwide survey. Br J Dermatol 2009; 161: 1153–1158.
- Nordenfelt P, Nilsson M, Björkander J, Mallbris L, Lindfors A, Wahlgren C. Hereditary angioedema in Swedish adults: report from the national cohort. Acta Derm Venereol 2016; 96: 540–545.
- Stray-Pedersen A. Primary immunodeficiency diseases in Norway. J Clin Immunol 2000; 20: 477–485.
- Laitinen I, Jokelainen J, Tasanen K, Huilaja L. Comorbidities of alopecia areata in Finland between 1987 and 2016. Acta Derm Venereol 2020; 100: adv00063.
- Heiskanen SL, Niskala J, Jokelainen J, Tasanen K, Huilaja L, Sinikumpu SP. Hyperhidrosis comorbidities and treatments: a register-based study among 511 subjects. Acta Derm Venereol 2022; 102: adv00656.
- Kurki M, Sinikumpu SP, Kiviniemi E, Jokelainen J, Huilaja L. Validation of diagnoses of atopic dermatitis in hospital registries: a cross-sectional database study from Finland. Acta Derm Venereol 2023; 103: adv7266.
- Haverinen S, Vihervaara A, Löyttyniemi E, Peltonen S, Koulu L, Tasanen K, et al. Validation of psoriasis diagnoses recorded in Finnish biobanks. Acta Derm Venereol 2020; 100: adv00297.
- Leisti P, Pankakoski A, Jokelainen J, Varpuluoma O, Huilaja L, Panelius J, et al. Accurate diagnosis of bullous pemphigoid requires multiple health care visits. Front Immunol 2023; 14: 1281302.