QUIZ SECTION
A 3-month-old Infant with Mottled Hypo- and Hyper-pigmented Patches on the Extremities: A Quiz
Suyeon KIM1, Dae-Hyun JANG2 and Hei Sung KIM1,*
1Department of Dermatology, 2Department of Rehabilitation Medicine, Incheon St Mary’s Hospital, College of Medicine, Catholic University of Korea, 222 Banpo-daero, Seocho-gu, Seoul 06591, Korea. *E-mail: hazelkimhoho@gmail.com
Citation: Acta Derm Venereol 2024; 104: adv26091. DOI https://doi.org/10.2340/actadv.v104.26091.
Copyright: © Published by Medical Journals Sweden, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/)
Published: Mar 14, 2024
A 3-month-old infant presented with mottled hypopigmented macules and patches with hyperpigmented dots and keratoderma on both the upper and lower extremities since birth (Fig. 1). Her parents denied any history of birth defects or perinatal events. Her father and grandmother were said to have similar pigmented findings since birth. There had been no evidence of systemic involvement.

Fig. 1. Skin features of the patients. Hypopigmented macules and patches with hyperpigmented dots on the extremities (A, B) and erythematous hyperkeratotic plaques and keratoderma (C).
Punch biopsies were performed from both the hypo- and hyperpigmented lesions. Histopathological examination showed a decrease in the basal layer melanin in the hypopigmented lesion and an increase in the melanin content of the hyperpigmented lesion (Fig. 2).

Fig. 2. Histologic findings. Hypopigmented lesion showing mild acanthosis and a decrease in melanin content in the basal layer (A). Hyperpigmented lesion showing mild acanthosis and an increase in melanin content of the basal layer (B). (A, B: H&E stain, x100).
What is your diagnosis?
Differential diagnosis 1: Cole disease
Differential diagnosis 2: Dyschromatosis symmetrica hereditaria (DSH, acropigmentation of Dohi)
Differential diagnosis 3: Reticulated acropigmentation of Kitamura (RAK)
Differential diagnosis 4: Epidermolysis bullosa with mottled pigmentation (EBS-MP)
See next page for answer.
ANSWERS TO QUIZ
A 3-month-old Infant with Mottled Hypo-and Hyper-pigmented Patches on the Extremities: A Commentary
Diagnosis: Cole disease
After informed consent and approval from the local institutional review board, we obtained clinical data and blood samples for next-generation sequencing (NGS) for participation in this report. Only the patient was genotyped for mutation (Fig. 3A). An NGS test for hereditary skin and pigmentary disorders was performed. For NGS, the peripheral blood sample was used for genomic DNA extraction and the TruSight One Extended Sequencing Panel Kit was used for sequencing on a NextSeq 550Dx instrument (Illumina, San Diego, CA). As a result, a heterozygous missense variant, G to A transition at nucleotide position 530 resulting in the substitution of cysteine(C) to arginine(R) (NM_006208.2:c.530G>A/p.(Cys177Tyr)), was revealed in the ENPP1 (Ectonuclotide pyrophosphatase/phosphodiesterase-1). This variant has not been previously reported in control databases. Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation Scores phyloP100: 7.613 and MetaRNN Score:0.994) and location in a mutational hot spot and/or critical and well-established functional domain without benign variation. This variant is suggested to be a “disease causing” variant, based on the American College of Medical Genetics and Genomics guidelines regarding the interpretation of sequence variations (PP3(strong)+PM1+PM2) (1). This mutation is included in 1 of the 7 reported ENPP1 mutations associated with Cole disease (Fig. 3B).
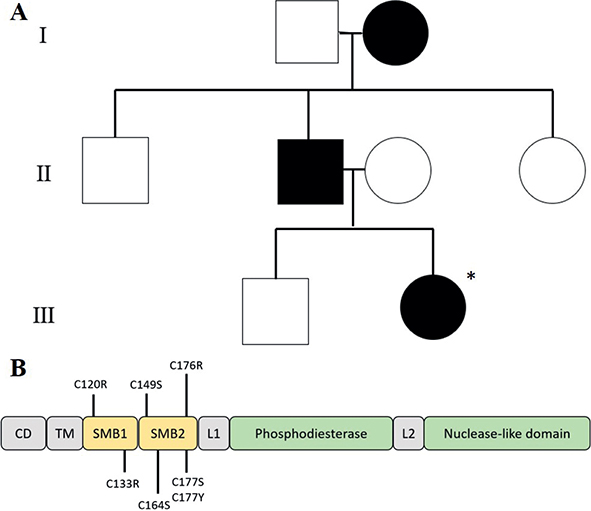
Fig. 3. Pedigree and domain organization of ENPP1 protein. (A) Pedigree of a family with Cole disease with autosomal dominant inheritance’ only the patient was genotyped for mutation in ENPP1 (marked with an asterisk). (B) Domain organization human ENPP1 protein with the localization of previously reported Cole disease mutation in the protein domains SMB1 and SMB2. CD, cytosolic domain, TM, transmembrane domain; L1, linker region1; L2, linker region2; SMB, somatomedin.
Based on clinicopathological features, the patient was diagnosed with Cole disease. To improve skin mottling, the patient was treated with a 311-nm excimer laser on the hypopigmented lesions once a week for six months. The parents reported subtle improvements. There has been no evidence of systemic involvement so far.
Cole disease (MIM 615522) is a rare hereditary genodermatosis, characterized by punctate keratoderma with irregularly shaped hypopigmented macules and rarely cutaneous calcifications. Since its first report in 1976 by Cole (2), 10 cases have been reported. Cole disease is known to show autosomal dominant inheritance. However, one report suggested Cole disease is an autosomal recessive disorder (3). Underlying ENPP1 mutation in Cole disease was first reported in 2013 (4).
The ENPP1 is composed of 8 domains, and ENPP1-associated genetic diseases can be classified by mutations in these locations. The phosphodiesterase domain, one of the domains in ENPP1, is known to inhibit ectopic calcifications and abnormal calcium handling (5). Generalized arterial calcification of infancy (GACI, MIM 208000), pseudoxanthoma elasticum (MIM 264800), and hypophosphatemic rickets type-2 (MIM 613312), which all include systemic involvement, are associated with this mutation. In contrast, somatomedin-B-like (SMB) 1 and 2 domains inhibit abnormal mineralization and insulin regulation (6). Insulin regulation is responsible for keratinocyte differentiation, epidermal growth factor signalling and melanosome transfer, which are associated with epidermal homeostasis (6). Cole disease patients have shown mutations in the SMB1 and 2 domains. Impaired insulin signalling is due to SMB1 and 2 mutation is likely associated with punctate palmoplantar keratoderma. Moreover, insulin regulates melanosome transfer from melanocytes to keratinocytes by the proteinase-activated receptor 2 (PAR-2) and mutation in this signalling causes a decrease in melanosome transfer (6). Systemic involvement associated with abnormal insulin regulation, such as diabetes, has not been reported in patients with Cole disease (7).
Cole disease typically presents as punctate keratoderma and mottled hypo- and hyperpigmentation on the extremities. Cutaneous calcifications have rarely been reported (8). Further study and follow-up are needed to clarify systemic involvement in Cole disease.
Cole disease shows non-specific histologic findings (9). Palmoplantar lesions typically show hyperkeratosis, hypergranulosis and acanthosis. The hypopigmented lesion shows a decrease in melanin contents of the basal layer with a normal number of melanocytes. This suggests the association of pathophysiology with only an interference in the melanosome transport, not a number of melanocytes (10).
Genedermatoses, which show mixed hyper- and hypopigmented macules, can be differentially diagnosed as dyschromatosis symmetrica hereditaria (DSH, acropigmentation of Dohi), reticulated acropigmentation of Kitamura (RAK) and epidermolysis bullosa with mottled pigmentation (EBS-MP) (7). These pigmentary disorders have similar characteristics, and genetic testing can be a useful tool to differentiate them.
Unfortunately, cutaneous manifestation normally persists for life without remission (6,7). However, as in our case, an excimer laser can be applied to hypopigmented lesions, and emollients should be applied continuously on the eczematous lesion. One patient with RAK was treated with a 532-nm Q-switched ND:YAG laser, which may also be a promising treatment for Cole disease by attenuating the hyperpigmented spots (8).
In conclusion, we report a child with Cole disease, a genodermatosis with a family history with involvement limited to the skin. Clinicians should always be aware of this rare hereditary dyschromatosis and perform gene studies to confirm the diagnosis.
ACKNOWLEDGEMENTS
This work was supported by the Catholic Medical Center Research Foundation made in the programme year of 2023.
Consent for publication of all patient photographs and medical information was provided by the authors at the time of article submission to the journal, stating that all patients gave consent for their photographs and medical information to be published in print and online and with the understanding that this information may be publicly available.
REFERENCES
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genet Med 2015; 17: 405–424.
- Cole LA. Hypopigmentation with punctate keratosis of the palms and soles. Arch Dermatol 1976; 112: 998–100.
- Chourabi M, Liew MS, Lim S, H’mida-Ben Brahim D, Boussofara L, Dai L, et al. ENPP1 mutation causes recessive Cole disease by altering melanogenesis. J Invest Dermatol 2018; 138: 291–300.
- Eytan O, Morice-Picard F, Sarig O, Ezzedine K, Isakov O, Li Q, et al. Cole disease results from mutations in ENPP1. Am J Hum Genet 2013; 93: 752–757.
- Staretz-Chacham O, Shukrun R, Barel O, Pode-Shakked B, Pleniceanu O, Anikster Y, et al. Novel homozygous ENPP1 mutation causes generalized arterial calcifications of infancy, thrombocytopenia, and cardiovascular and central nervous system syndrome. Am J Med Genet A 2019; 179: 2112–2118.
- Gabaton N, Kannu P, Pope E, Shugar A, Lara-Corrales I. A novel ENPP1 mutation identified in a multigenerational family affected by Cole disease. Pediatr Dermatol 2020; 37: 868–871.
- Li Z, Wang L, Wang S. Hyperpigmentation in a Chinese family with autosomal dominant Cole disease. Exp Dermatol 2022; 31: 248–254.
- Lee JH, Lee JH, Lee JH. A case of reticulate acropigmentation of Kitamura treated with 532-nm Q-switched Nd:YAG laser: 10 years of follow-up observation. Ann Dermatol 2014; 26: 783–785.
- Vignale R, Yusín A, Panuncio A, Abulafia J, Reyno Z, Vaglio A. Cole disease: hypopigmentation with punctate keratosis of the palms and soles. Pediatr Dermatol 2002; 19: 302–306.
- Moore MM, Orlow SJ, Kamino H, Wang N, Schaffer JV. Cole disease: guttate hypopigmentation and punctate palmoplantar keratoderma. Arch Dermatol 2009; 145: 495–497.