Livedoid Pattern in the Abdomen of a Patient with Myelofibrosis: A Quiz
Diego Soto-García1,2, Gonzalo Peón1,2, Carlos Álvarez3, Carmen Couselo-Rodríguez1,2 and Ángelez Flórez1,2
1DIPO Research Group, Galicia Sur Health Research Institute (IIS Galicia Sur), SERGAS-UVIGO, Vigo, 2Department of Dermatology and 3Department of Pathology, University Hospital of Pontevedra, Av. Montecelo, ES-36164 Casas Novas, Pontevedra, Spain. E-mail: diego.soto.garcia@gmail.com
doi: 10.2340/actadv.v101.277
A 78-year-old man presented to Department of Dermatology at University Hospital, Pontevedra, with a 3-month history of a painful eruption on the lower trunk and groin. The lesions had been treated previously with topical clobetasol, with no response. The patient did not report systemic symptoms, and had not taken any new medications over the preceding weeks.
Regarding previous medical history, the patient had been diagnosed with JAK2-positive myelofibrosis 6 years previously and was receiving ruxolitinib, hydroxyurea, aspirin and erythropoietin to control the disease. To date, he remains stable in this regard.
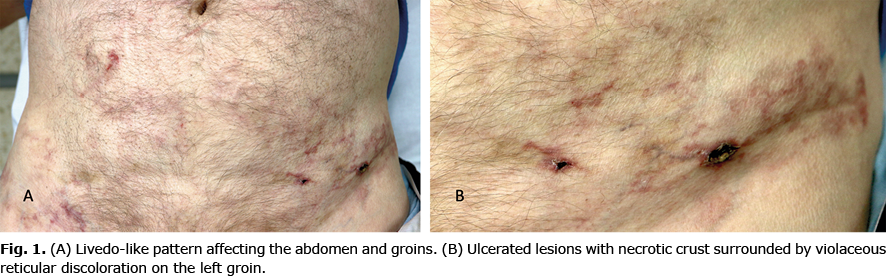
Clinical examination revealed a discontinuous reticular and violaceous macular discolouration, purpuric and ulcerated lesions affecting his trunk and groins (Fig. 1). Blood test revealed an abnormal haemogram with haemoglobin 10.5 g/dl (normal 13.5–17.5 g/dl), leucocytes 27.4 ×109/l (normal 4.5–11×109/l), neutrophils 22.6×109/l (normal 1.8–8×109/l), platelets 108 ×109/l (normal 130–400×109/l); and elevated creatine levels 1.78 mg/dl (normal 0.7–1.2 mg/dl). Serum antinuclear antibodies were negative and anti-neutrophil cytoplasmic autoantibodies were positive at 1/20 title (p-ANCA); complement C3 and C4 levels were normal. Cryoglobulins were also absent and serum proteinogram did not show notable alterations. All the serology tests (including HIV, hepatitis C and hepatitis B) and all the skin tissue cultures (including bacteria, fungus and mycobacteria) were negative. A punch biopsy was performed and sent for histopathological evaluation (Fig. 2).

What is your diagnosis? See next page for answer.
Livedoid Pattern in the Abdomen of a Patient with Myelofibrosis: A Commentary
Acta Derm Venereol 2022; 102: adv00623.
Diagnosis: Reactive angioendotheliomatosis
Histopathological examination of the skin biopsy performed on the abdomen revealed a benign intraluminal proliferation of endothelial cells in tightly packed and compressed capillaries varying in size, associated mild chronic inflammatory infiltrate and increased number of capillary-sized blood vessels in the dermis, forming lobules. These pathological features confirmed the diagnosis of reactive angioendotheliomatosis (RAE). RAE is a rare benign angioproliferative condition of the skin, which has been noted to occur in patients with a variety of underlying systemic and inflammatory diseases, including also end-stage renal disease, autoimmune diseases, hypercoagulable conditions, malignancies, such as lymphoproliferative diseases, cryoproteinaemia, amyloid endocarditis, and infective endocarditis (1–5). Prothrombotic disorders are the more frequently reported associated with RAE (5). RAE may present with a wide range of clinical findings, such as erythemato-violaceous or purpuric macules, livedoid pattern, papules, nodules or plaques, of very different sizes and number of lesions. Involved areas may be painful, pruritic, or asymptomatic (1).
Histopathologically, this condition is characterized by a proliferation of capillaries that predominantly involve the dermis. Lesions are generally poorly marginated and heterogeneous between cases, and often also between lesions in an individual patient (1). At higher magnification, lesions are composed predominantly of a proliferation of closely packed capillaries that lack significant endothelial atypia. In most cases there is an associated mild chronic inflammatory infiltrate that is generally lymphocytic or lymphoplasmacytic. Other features include scattered fibrin Microthrombi, which often show reactive fasciitis-like dermal alterations and red cell extravasation. Occasional mitotic figures in the endothelium can be commonly seen. The intravascular cellular proliferation stains positively for CD31 and CD34 (1, 6).
The pathogenesis of RAE is unknown. Indeed, the more varied the coexistent systemic diseases, the more difficult it is to suggest a unifying pathogenetic process. Vessel occlusion and tissue hypoxia have been proposed as one of the common pathological mechanisms underlying the reactive vascular and endothelial proliferative changes observed in these patients who develop RAE (1, 2, 7). The current patient had a history of myelofibrosis, which is a well-known prothrombotic disease and its traditional therapy is the prevention of thrombotic events with antiproliferative agents (in the current patient ruxolitinib and hydroxyurea) in association with aspirin (8, 9). We consider that in the current patient the prothrombotic environment due to myelofibrosis was the main underlying cause for the appearance of RAE.
Differential clinical diagnoses include vascular tumours, such as angiosarcoma and Kaposi sarcoma, and diseases that may present with livedo racemose, such as polyarteritis nodosa or thrombophlebitis (1, 2). Histopathologically, other intravascular proliferations must be ruled out, such as intravascular papillary endothelial hyperplasia, intravascular pyogenic granuloma, intralymphatic histiocytosis (which cells are CD68 positive) and angiotropic lymphoma (1, 6). Angiotropic lymphoma was historically referred to as malignant angioendotheliomatosis; it is now more properly known as intravascular large cell lymphoma or angiotropic lymphoma. The misnomer angioendotheliomatosis stems from the fact that this condition is characterized by an intraluminal proliferation of large atypical cells that were thought to be derived from endothelial cells. Advances in immunohistochemistry demonstrated that these neoplastic cells are of lymphoid origin, and while cases of T-cell origin have been reported, the majority of cases are B-cell proliferations (6, 10, 11).
RAE does not currently have a specific treatment. Regarding therapy, we want to highlight the importance of the evaluation of possible known associations because lesions can remit if the underlying cause is solved (e.g. revascularization of ischaemic territories).
In conclusion, RAE may be a potential harbinger of systemic disease, as this association appeared in approximately 75% of the RAE-reported cases (1). Therefore, clinical investigation and follow-up should be considered in a case of RAE arising in the absence of any apparent underlying condition.
The patient provided written consent for the use of his photographs and medical records.
The authors have no conflicts of interest to declare.
REFERENCES
- McMenamin MC, Fletcher CD. Reactive angioendotheliomatosis a study of 15 cases demonstrating a wide clinicopathologic spectrum. Am J Surg Pathol 2002; 26: 685–697.
- Singer C, Mallon D, Auguston B, Lam M, Foster R. Reactive angioendotheliomatosis presenting as livedo racemosa secondary to propylthiouracil. Pathology 2020; 52: 494–496.
- Lazova R, Slater C, Scott G. Reactive angioendotheliomatosis. Case report and review of the literature. Am J Dermatopathol 1996; 18: 63–69.
- Thai K, Barrett W, Kossard S. Reactive angioendotheliomatosis in the setting of antiphospholipid syndrome. Australas J Dermatol 2010; 44: 151–155.
- Di Filippo Y, Cardot-Leccia N, Long-Mira E, Andreani M, Richez V, Lacour JP, et al. Reactive angioendotheliomatosis revealing a glomerulopathy secondary to a monoclonal gammopathy successfully treated with lenalidomide. J Eur Acad Dermatol Venereol 2021; 35: 115–118.
- Mazloom SE, Stallings A, Kyei A. differentiating intralymphatic histiocytosis, intravascular histiocytosis, and subtypes of reactive angioendotheliomatosis: review of clinical and histologic features of all cases reported to date. Am J Dermatopathol 2017; 39: 33–39.
- Kirke S, Angus B, Kesteven PJ, Calonje E, Simpson N. Localized reactive angioendotheliomatosis. Clin Exp Dermatol 2007; 32: 45–47.
- Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012; 366: 787–798.
- Gangat N, Tefferi A. Myelofibrosis biology and contemporary management. Br J Haematol 2020; 191: 152–170.
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks. Blood 2018; 132: 1561–1567.
- Perniciaro C, Winkelmann RK, Daoud MS, Su WP. Malignant angioendotheliomatosis is an angiotropic intravascular lymphoma. Immunohistochemical, ultrastructural, and molecular genetics studies. Am J Dermatopathol 1995; 17: 242–248.