Segmental pigmentation disorder (SPD) is characterized by hypo- or hyper-pigmented patches segmentally distributed, present in infancy, more prominently in darker-skinned children. The aim of this study was to define the demographic and clinical characteristics of SPD in a large series of patients. This was a retrospective case-control study at 2 paediatric dermatology centres in Israel. Data were collected through a telephone questionnaire and medical records. The study group consisted of 144 individuals with SPD and 144 individuals visiting the same institutions matched for age and sex. Median age of onset of SPD was near birth; 51% of patients were Sephardic Jews, and patients were followed up for a median period of 27 years. The patches were located on the torso (43%), mostly hypopigmented (52%), and remained of the same intensity and size in 55% and 41% of cases, accordingly. No differences in extracutaneous morbidities were found between SPD and control patients. This study delineates the demographic and clinical characteristics of SPD, confirms that cutaneous findings in SPD are more prominent in darker skin, tends not to expand in size or accentuate throughout the years, nor to be associated with extracutaneous morbidities.
Key words: pigmentation; segmental; mosaicism; extracutaneous manifestations.
Accepted Mar 21, 2022; Epub ahead of print Mar 21, 2022
Acta Derm Venereol 2022; 102: adv00707.
DOI: 10.2340/actadv.v102.399
Corr: Ilan Goldberg, Department of Dermatology, Tel Aviv Sourasky Medical Center. 6 Weizman Street, Tel Aviv 64239, Israel. E-mail: ilangoldberg1@gmail.com
SIGNIFICANCE
This study supports the assumption of segmental pigmentation disorder being a solely cutaneous disorder, without any obligatory association of extracutaneous manifestations, which may diminish the need for long-term neurological follow-up and alleviate the burden felt by the child’s parents when informed of the diagnosis and its possible outcomes.
INTRODUCTION
In 1983, the term segmental pigmentation disorder (SPD) was first introduced by Metzker (1), referring to a phenomenon prevalent among dark-skinned children, consisting of hypo- or hyper-pigmented patches and featuring a segmental distribution with a sharp midline frontal delineation. In the medical literature, this phenomenon was depicted under many names, including “segmental pigmentation anomaly”’, “segmental naevus depigmentosus”, “giant café-au-lait macule”, “patterned dyspigmentation”, “pigmentary mosaicism” and more (2–5). In 2010 Hogeling & Frieden (6) further broadened the definition of the term SPD, to include the information that the pigmented patch can assume a segmental, dermatomal or chequerboard pattern (7).
Several factors have been suggested to account for the unique appearance of SPD, including the effect of neural and hormonal factors on pigment cells, somatic mosaicism and cellular chimerism (7–10).
Skin pigmentation develops in a mediolateral direction and does not cross the linea alba. Aberrant pigment dispersion is expected to lead to an irregular excess or lack of pigment, for the most part only on 1 side, a fact which may explain the sharp midline border typical of SPD (1).
SPD is usually sporadic, although familial cases have been reported (1, 6). Paradominant inheritance may explain familial cases: an individual heterozygous for a paradominant mutation has a normal phenotype, thus the mutant allele can be passed on to future generations without being phenotypically discernible, unless a somatic mutation appears at an early stage of embryonic development, causing loss of heterozygosity and forming a homozygous subpopulation of cells. This results in a mosaic phenotype comprising 2 subsets of cell populations; homo and heterozygous. Once the skin development in the foetus is completed, paradominant features can no longer be expressed (7).
SPD appears during infancy and is characterized by a small number of lesions distributed segmentally, typically involving the frontal torso. The midline border is predominantly frontal and sharp, and the lateral border is less salient. The SPD lesions may cross the midline; however, by no more than a few centimetres (6) and tend to fade over time (1) (although pigmentation may persist into adulthood; 2, 6). SPD has not been found to be convincingly associated with extracutaneous manifestations (1, 6). Atrial septal defect (ASD), strabismus with retinal hypopigmentation, and bronchogenic cyst have been reported in patients with SPD and other mosaic-patterned pigmentation disorders, but it is not clear to what extent this association reflects a causal relationship (6, 11).
Hyperpigmented SPD lesions are characterized histologically by a higher amount of melanin than usual in the stratum basale due to an increased production (1), similarly to histological findings reported in café au lait spots (12). Hypopigmented lesions show less melanin than usual, but the number of melanocytes is normal or reduced (13–15).
The differential diagnosis of hyperpigmented lesions include giant café-au-lait macules (2, 3, 6, 16), McCune-Albright syndrome (17–19), speckled lentiginous naevi (20), naevoid hyperpigmentation (6, 21), congenital pigmented naevus (22–26), congenital giant Becker’s naevus (27, 28) and segmental neurofibromatosis (2, 29). The differential diagnosis of hypopigmentated SPD (30) spans segmental vitiligo (6), naevus anaemicus (21), tuberous sclerosis (32) and naevoid hypopigmentation, such as hypomelanosis of Ito (11, 21).
SPD is likely to be more common than reported in the literature.
The aim of this study was to systematically characterize the clinical features of SPD in a large cohort of affected children to establish whether the diagnosis of SPD requires further medical investigations or follow-up.
MATERIALS AND METHODS
Study population
This was a retrospective case-control study. A total of 185 subjects were diagnosed with SPD at 2 medical centres in Israel: 145 patients who were evaluated at the Children’s Dermatology Clinic at Schneider Medical Center between the years 1975 and 1995, and 40 patients evaluated at the Tel-Aviv Sourasky Medical Center between 1995 and 2012. SPD was defined as a hypo- or hyper-pigmented patch in a segmental distribution with a well-defined midline demarcation first distinguished in early infancy. Patients with other defined pigmentary disorders were excluded, such as giant café-au-lait macules (round or oval patches, which lack the characteristic midline demarcation and segmental distribution) (2, 3, 6, 16), McCune-Albright syndrome (multiple hyperpigmented macules/patches following Blaschko’s lines with additional characteristic systemic manifestations as precocious puberty and polyostotic fibrous dysplasia) (17–19), naevoid hyperpigmentation (multiple whorled hyperpigmented patches along Blaschko’s lines) (6, 21), segmental neurofibromatosis (discrete oval or round hyperpigmented macules expressed with additional findings, as axillary and intertriginous freckling and neurofibromas) (2, 29), segmental vitiligo (depigmented well-defined macules) (6), etc.
The control group consisted of subjects who visited the 2 centres during the same years because of other skin disorders, not associated with neurological manifestations, including atopic dermatitis, hyperhidrosis, keratosis pilaris, dyshidrotic eczema, and seborrhoeic dermatitis. The subjects were paired according to age and sex.
Data were collected from the patients’ digital medical records, handwritten medical charts, photographed slides of physical findings at baseline (enabling further clinical description of the pigmentation characteristics at time of diagnosis) and a phone interview. Data collected included demographic details (sex, age at presentation and at follow-up); ethnic background (ethnic origin was defined based on the origin of 2 or more grandparents); clinical details including characteristics and changes in size over time of the pigmented patch (based on medical records, charts, interviews and photographs), family history, additional cutaneous and extracutaneous manifestations, and evaluation of personal and social coping extracted from the telephone questionnaire.
Data analysis
Continuous variables were described using a mean and standard deviation (SD); continuous variables that did not fit a normal distribution were described using a median and interquartile range (IQR). Categorical variables were presented using frequency and percentages. A univariate analysis was used to compare the demographic data, the additional skin phenomena, the family history, and the existence of extracutaneous manifestations. The Pearson χ2 test and the Fisher’s exact test were used to compare the categorical variables between the study and control groups. The 2-sample Wilcoxon test was used to compare the continuous variables among the 2 groups. When the results of all the tests for categorical variables that consisted of more than 3 categories were significant, pairwise comparisons were performed. The false discovery rate method was used to match the level of significance. Differences were determined as significant when the p-value was less than 0.05. The statistical analysis of the data was performed using SAS system for Windows (SAS Institute Inc., Cary, NC, USA).
RESULTS
A total of 185 subjects were diagnosed with SPD, of which 144 individuals had accessible medical records, thus comprising the study group; the control group consisted of 144 subjects diagnosed with atopic dermatitis (n = 112, 78%), bullous impetigo (n = 13, 9%), hyperhidrosis (n = 15, 10%), keratosis pilaris, dyshidrotic eczema, seborrhoeic dermatitis and molluscum contagiosum (1 of each, total 3%).
Comparative demographic data are shown in Table I. The groups were paired according to age and sex (p-value not indicating variance for both). Variance was ascertained based on age at examination and ethnic origin (a statistically significant p-value). Most patients were adults 25–42 years (76% in each group), females (60%) and of Sephardic Jewish origin. The median age of onset of SPD is close to birth, with a mean of 1.33 years.
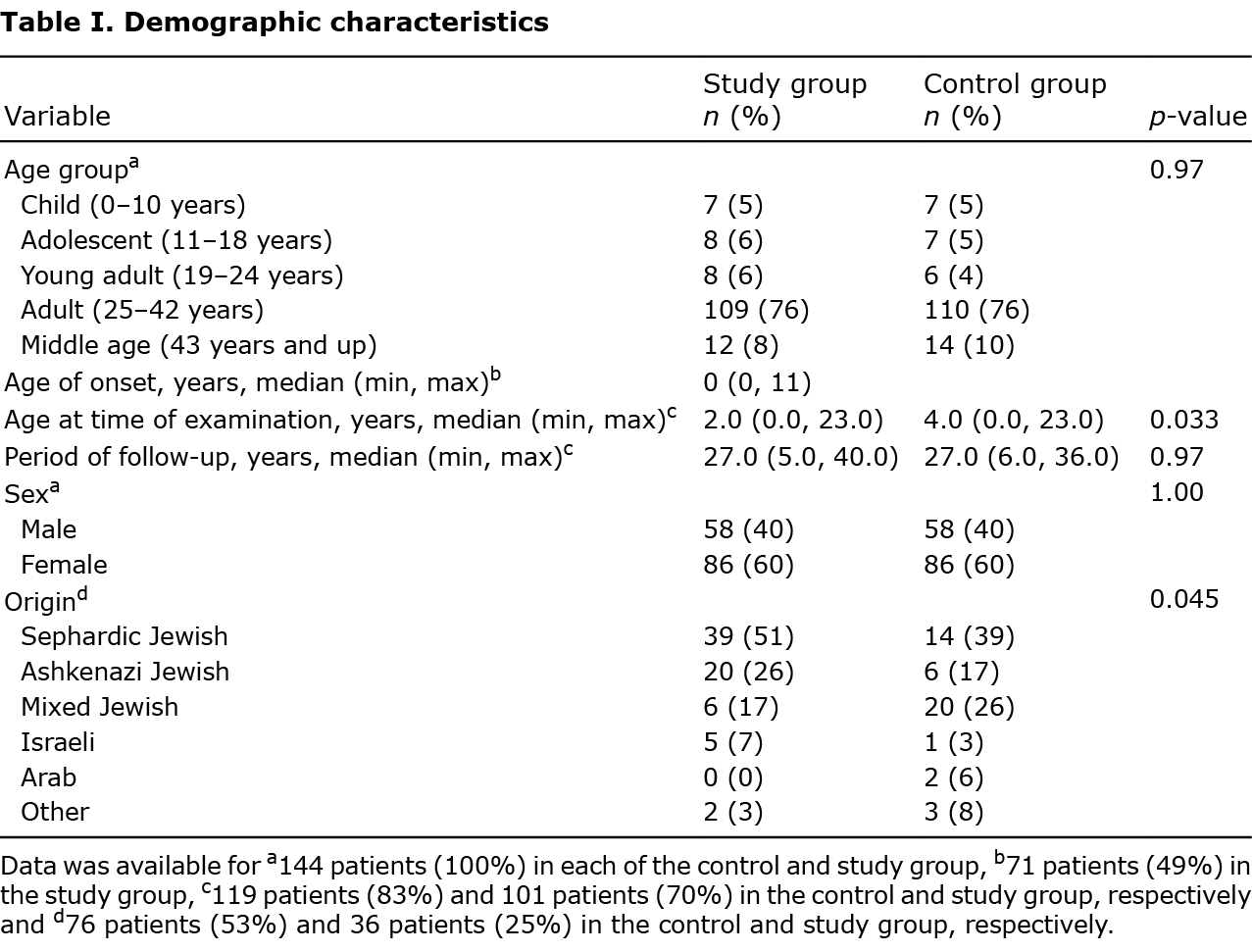
Table II presents the clinical characteristics of the pigmentation. Most of the pigmentary lesions were distributed across several anatomical locations (44%, most often with torso involvement) or located solely on the torso (43%), and were hypopigmented patches (52%), with a frontal demarcation midline (61%) and no clear predilection to ill- (48%) or well-defined (47%) borders (Fig. 1). Throughout the years the pigmentation size and intensity remained unchanged in 41% and 55% of cases, respectively. Ninety-five percent of SPD patients did develop additional cutaneous manifestations. Most SPD patients did not have a first-degree relative diagnosed with SPD (89%), though 2 patients had a sibling with SPD (7%) and 1 affected individual had a parent diagnosed with SPD (4%).

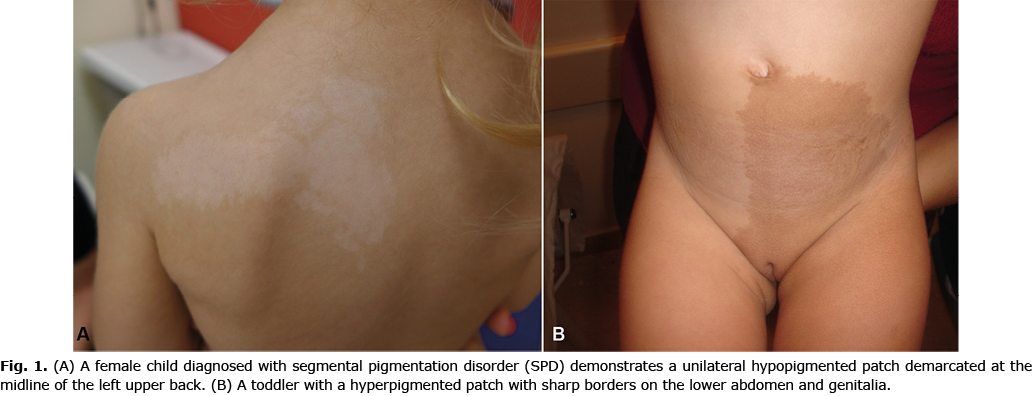
SPD was asymptomatic in all patients. Data regarding personal coping were based on 28 patients with up-to-date contact details who completed a telephone questionnaire. Most SPD patients did not report any aesthetic concern (64%) or impairment of quality of life (89%), whereas half of the subjects reported experiencing social consequences, such as negative remarks and a sense of embarrassment.
No differences in family medical history, personal morbidity, medications and neurological manifestations were found between the 2 groups. Family history and co-morbidities among patients with SPD and control individuals are listed in Table SI.
DISCUSSION
This study of a large cohort of patients comprehensively delineated the demographic, cutaneous and extracutaneous manifestations of SPD over a long period of follow-up. Table III compares the principal findings of the current study with the available literature. SPD is first expressed near birth (median age 0); previous studies suggested that that the pigmentation appears slightly after birth, but is not evident immediately at birth, due to the infant’s skin’s initial predisposition to hypopigmentation and its lack of exposure to the sun (13, 14). Consistent with previous data (1, 6), the current study could not identify any sex predominance.
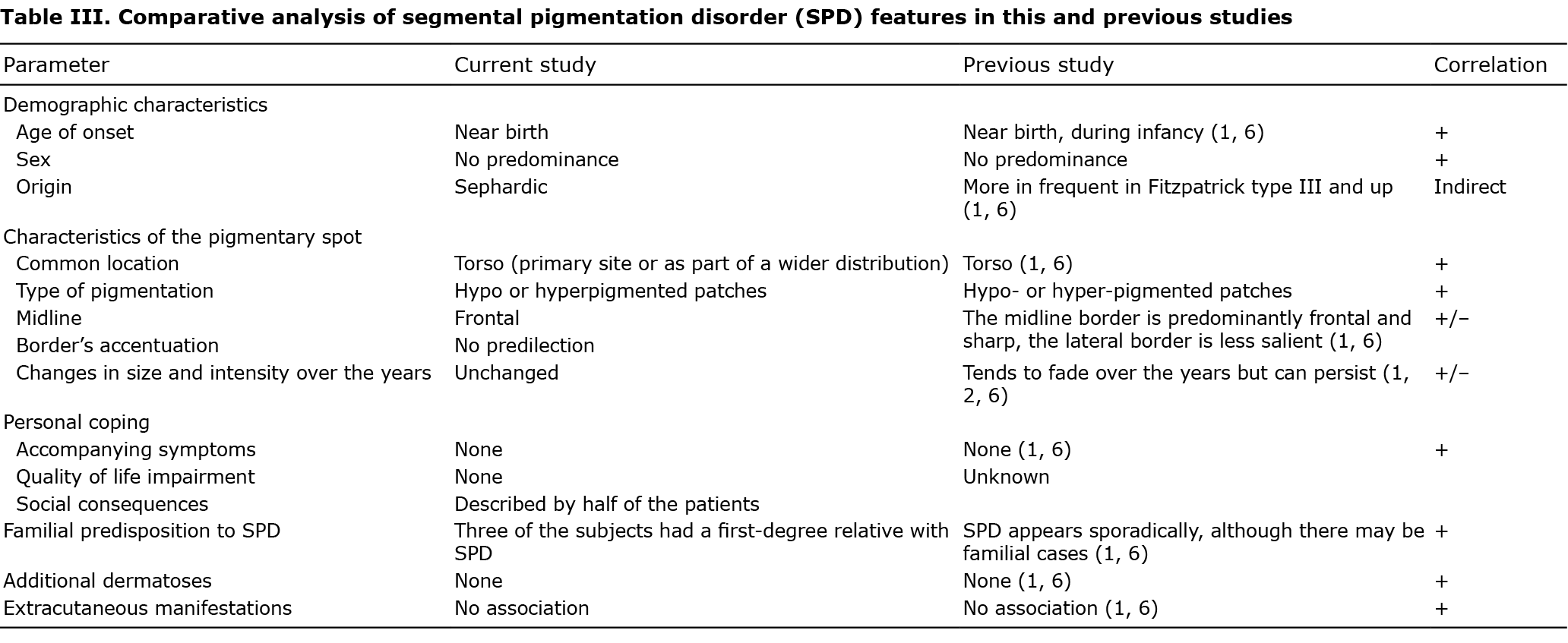
SPD tends to manifest in children with dark skin (1, 6). This may be consistent with the fact that most of the patients in the current series were of Sephardic origin, as reported in a previous study (1).
The current study shows that SPD commonly involves the torso. This finding is in agreement with the observations of both Metzker (1) and Hogeling & Frieden (6), who found that the most common locations of SPD in an ascending order were the back, chest, and abdomen.
Slightly more than half of the patients had hypopigmentary patches (52%). Similarly, Metzker (1) found that 50% of subjects with SPD display hypopigmented lesions. In contrast, Hogeling & Frieden (6) report hyperpigmentation in 77% of patients with SPD.
The current study found no significant differences regarding ill- vs well-defined borders. The lesion midline border was mostly frontal; Hogeling & Frieden (6) also found a frontal midline in most of their patients (82%).
Previous studies showed that pigmentation tends to fade over the years (1), but could persist throughout adulthood. Hogeling & Frieden (6) described 2 adults with SPD. Orion et al. (2) described a series of cases in which the pigmentation did not fade over time. The current study suggests that pigmentation intensity or size do not change over the years.
In accordance with the literature, SPD was found to be asymptomatic (1, 6). In addition, the psychological impact of SPD was ascertained here for the first time to the best of our knowledge. No significant deleterious effect was observed, although half of the subjects experienced negative social remarks.
A first-degree relative with SPD was found in only 3 cases (11%), which could be explained by the mechanism of paradominant inheritance (33).
Extracutaneous findings have been related in the past to mosaic-patterned pigmentation disorders, such as hypomelanosis of Ito and linear and whorled hypermelanosis, including neural, ocular and cardiac manifestations (11). Similarly, cases of naevus depigmentosus accompanied by disorders of the central nervous system were described: Kim et al. (14) described a case of localized naevus depigmentosus with seizures (1 out of 60 subjects), out of which 32 patients had a segmental picture that may matched SPD, and Nehal et al. (36) described one subject with systemic manifestations out of 9 with segmental naevus depigmentosus. In most cases of naevus depigmentosus there are no systemic extracutaneous manifestations, and, in particular, no neurological manifestations (11, 14, 15, 37). In the current study, most of the subjects were healthy, and co-morbidities were not significantly associated with SPD, as previously found (6).
Study limitations
This study was limited by its retrospective design. In addition, some of the data had been collected through a questionnaire, suggesting the possibility of a recall bias. Data regarding personal consequences of the pigmentary disorder were based solely on the telephone questionnaire fulfilled by 20% of patients, thus withholding an incomplete data bias.
Conclusion
SPD tends to manifest during the first year of life, it is more common among children of Sephardic origin, is rarely familial, and consists of hypo- or hyper-pigmented asymptomatic patches, most often located on the torso, with a frontal midline, which can persist into adulthood. SPD is not associated with extracutaneous manifestations; an observation that carries obvious implications for the counselling of patients with SPD and their families.
The authors have no conflicts of interest to declare.
REFERENCES
- Metzker A, Morag C, Weitz R. Segmental pigmentation disorder. Acta Derm Venereol 1983; 63: 167–169.
- Orion E, Matz H, Wolf D, Wolf R. Café au lait has a hue of its own. Dermatol Online J 2003; 9: 8.
- Min EA. Puzzling brown area on toddler’s skin. JAAPA 2007; 20: 15.
- Lombillo VA, Sybert VP. Mosaicism in cutaneous pigmentation. Curr Opin Pediatrics 2005; 17: 494–500.
- Taïeb A, Boralevi F. Hypermelanoses of the newborn and of the infant. Dermatol Clin 2007; 25: 327–336.
- Hogeling M, Frieden IJ. Segmental pigmentation disorder. Br J Dermatol 2010; 162: 1337–1341.
- Happle R. Mosaicism in human skin. Understanding the patterns and mechanisms. Arch Dermatol 1993; 129: 1460–1470.
- Lerner AB, Dnell RS, Chanco-turner ML, McGuire JS. Vitiligo and sympathectomy. Arch Dermatol 1966; 94: 269–277.
- Cho E, Cho SH, Lee JD. Patterned pigmentation in a child: a case of segmental pigmentation disorder. J Dermatol 2011; 38:1094–1096.
- Findlay GH, Moores PP. Pigment anomalies of the skin in the human chimaera, their relation to systematized naevi. Br J Dermatol 1980; 103: 489–498.
- Loomis CA. Linear hypopigmentation and hyperpigmentation, including mosaicism. Semin Cutan Med Surg 1997; 16: 44–53.
- Ortonne JP, Brocard E, Floret D, Perrot H, Thivolet J. Diagnostic value of café-au-lait spots. Ann Dermatol Venereol 1980; 107: 313–327.
- Lee HS, Chun YS, Hann SK. Nevus depigmentosus: clinical features and histopathologic characteristics in 67 patients. J Am Acad Dermatol 1999; 40: 21–26.
- Kim SK, Kang HY, Lee ES, Kim YC. Clinical and histopathologic characteristics of nevus depigmentosus. J Am Acad Dermatol 2006; 55: 423–428.
- Xu AE, Huang B, Li YW, Wang P, Shen H. Clinical, histopathological and ultrastructural characteristics of naevus depigmentosus. Clin Exp Dermatol 2008; 33: 400–405.
- Landau M, Krafchik BR. The diagnostic value of café-au-lait macules. J Am Acad Dermatol 1999; 40: 877–890.
- Rieger E, Kofler R, Borkenstein M, Schwingshandl J, Soyer HP, Kerl H. Melanotic macules following Blaschko’s lines in McCune–Albright syndrome. Br J Dermatol 1994; 130: 215–220.
- Völkl TM, Dörr HG. McCune–Albright syndrome: clinical picture and natural history in children and adolescents. J Pediatr Endocrinology Metab 2006; 19: 551–559.
- Davies JH, Barton JS, Gregory JW, Mills C. Infantile McCune–Albright syndrome. Pediatr Dermatol 2001; 18: 504–506.
- Schaffer JV, Orlow SJ, Lazova R, Bolognia JL. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol 2001; 137: 172–178.
- Kalter DC, Griffiths WA, Atherton DJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol 1988; 19: 1037–1044.
- Sarma N. Pigmentary nevi on face have unique patterns and implications: the concept of Blaschko’s lines for pigmentary nevi. Indian J Dermatol 2012; 57: 30–34.
- Effendy I, Happle R. Linear arrangement of multiple congenital melanocytic nevi. J Am Acad Dermatol 1992; 27: 853–854.
- Langenbach N, Pfau A, Landthaler M, Stolz W. Naevi spili, Café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol 1998; 78: 378–380.
- Nachbar F, Merkle T, Ruzicka T, Plewig G. Congenital junctional naevi following Blaschko’s lines. Eur J Dermatol 1993; 3: 478–479.
- Hanayama H, Terashi H, Hashikawa K, Tahara S. Congenital melanocytic nevi and nevus spilus have a tendency to follow the lines of Blaschko: an examination of 200 cases. J Dermatol 2007; 34: 159–163.
- Rao AG. Bilateral symmetrical congenital giant Becker’s nevus: a rare presentation. Indian J Dermatol 2015; 60: 522.
- Sood A, D’Souza P, Verma KK. Becker’s naevus occurring at birth and in early childhood. Acta Derm Venereol 1998; 78: 311.
- Listernick R, Mancini AJ, Charrow J. Segmental neurofibromatosis in childhood. Am J Med Genet 2003; 121A: 132–135.
- Bolognia JL, Pawelek JM. Biology of hypopigmentation. J Am Acad Dermatol 1988; 19: 217–255.
- Akhami RN, Schwartz RA. Nevus anemicus. Dermatology 1999; 198: 327–329.
- Trauner MA, Ruben BS, Lynch PJ. Segmental tuberous sclerosis presenting as unilateral facial angiofibromas. J Am Acad Dermatol 2003; 49: S164–166.
- Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol 1987; 16: 899–906.
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology, 3rd edn. Elsevier, China; 2012: p. 203–204.
- Little H, Kamat D, Sivaswamy L. Common neurocutaneous syndromes. Pediatric Annals 2015; 44: 496–504.
- Nehal KS, PeBenito R, Orlow SJ. Analysis of 54 cases of hypopigmentation and hyperpigmentation along the lines of Blaschko. Arch Dermatol 1996; 32: 1167–1170.
- Dhar S, Kanwar AJ, Kaur S. Nevus depigmentosus in India: experience with 50 patients. Pediatr Dermatol 1993; 10: 299–300.