QUIZ SECTION
A 71-year-old Woman with CREST Syndrome and Multiple Waxy Facial Papules and Plaques: A Quiz
Nidia PLANELLA-FONTANILLAS1,2, David PESQUÉ1,2, Laura PARRA-NAVARRO1,2 and Ramon M. PUJOL1,3
1Department of Dermatology, Hospital del Mar, Passeig Marítim, 25-29, ES-08003 Barcelona, 2Universitat Autònoma de Barcelona (UAB), Barcelona, and 3Universitat Pompeu Fabra (UPF), Barcelona, Spain. E-mail: nidia.planella.fontanillas@psmar.cat
Citation: Acta Derm Venereol 2024; 104: adv40419. DOI: https://doi.org/10.2340/actadv.v104.40419.
Copyright: 2024 © The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Published: Jun 8, 2024
A 71-year-old woman with a history of bronchiectasis and localized systemic sclerosis (CREST syndrome), manifested as Raynaud’s phenomenon, sclerodactyly, pathological capillaroscopy, and positive anti-centromere antibodies was referred for the evaluation of progressively growing facial papules and plaques over the last 6 months. Physical examination revealed 5 to 10 shiny, waxy, yellow-orange non-confluent asymptomatic papules and plaques measuring 0.5–2 cm in diameter on the left frontal area and right upper eyelid (Fig. 1). Laboratory results, encompassing complete blood cell count, were normal. Total serum protein and electrophoresis showed no abnormalities, and no paraprotein was detected. Additionally, roentgenographic studies, including chest and abdominal radiographs and echocardiogram, along with abdominal fat fine-needle aspiration, yielded normal results. A skin biopsy was performed, shown in Fig. 2. In-situ hybridization for immunoglobulin light chains revealed a monotypic expression pattern of lambda light chain. Polymerase chain reaction demonstrated clonal rearrangement of the immunoglobulin heavy chain gene (CDRII).
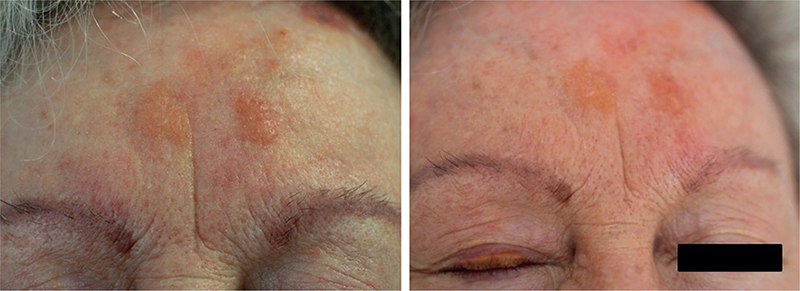
Fig. 1. Shiny, waxy, yellow-orange non-confluent asymptomatic papules and plaques on the left frontal area and right upper eyelid. Written informed consent for these photos was obtained.
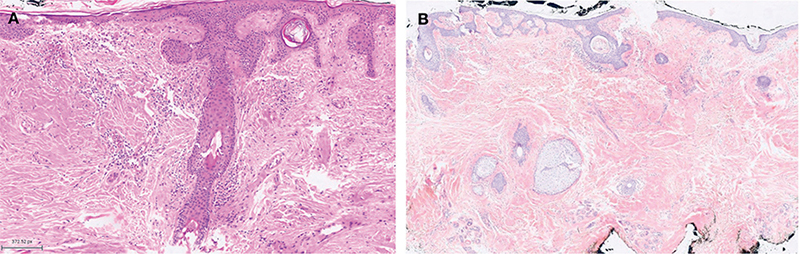
Fig. 2. (A) Haematoxylin and eosin stain (H/E) ×10: deposits of amorphous, eosinophilic, hyaline material in the superficial and mid-dermis primarily surrounding vascular and adnexal structures and discrete perivascular inflammatory infiltrate containing plasma cells is observed. (B) Deposits with Congo Red staining.
What is your diagnosis?
Differential diagnosis 1: Primary localized cutaneous nodular amyloidosis
Differential diagnosis 2: Lipoid proteinosis
Differential diagnosis 3: Colloid milium
Differential diagnosis 4: Erythropoietic protoporphyria
See next page for answer.
ANSWERS TO QUIZ
A 71-year-old woman with CREST Syndrome and Multiple Waxy Facial Papules and Plaques: A Commentary
Diagnosis: Primary localized cutaneous nodular amyloidosis associated with CREST syndrome
Primary localized cutaneous nodular amyloidosis (PLCNA) is a rare subset of cutaneous localized amyloidosis. Clinically, it presents as asymptomatic yellow-brown nodules or plaques, typically ranging from 0.5 to 7 cm in diameter, with a predilection for acral sites. More frequently observed in women during their fifth or sixth decade, these nodules may display telangiectasias, purpura, and, rarely, ulceration. The surrounding skin may appear atrophic (1). Histopathologically, PLCNA typically reveals diffuse hyaline amyloid deposits in the dermis, occasionally extending to the subcutaneous tissue and often surrounding blood vessels – a key diagnostic feature. The hyaline material becomes more evident with Congo Red staining (Fig. 2B), showing a green birefringence when examined under polarized light. In contrast to keratin-derived localized cutaneous amyloidosis types (macular, lichenoid, biphasic, dyschromic, and macular familial amyloidosis), amyloid deposits in PLCNA are not confined to the papillary dermis. They consist of immunoglobulin Lambda (most common) or Kappa light chains, referred to as amyloid L (AL) type. Importantly, PLCNA does not meet the criteria for systemic amyloidosis or hematologic dyscrasias (2).
Histologically, the differential diagnosis should consider other disorders presenting eosinophilic dermal deposits such as lipoid proteinosis, colloid milium, and erythropoietic protoporphyria. However, the morphological features, distribution, and Congo red positive staining of amyloid material aid in distinguishing this condition from others.
The potential risk of progression of PLCNA to systemic amyloidosis has been estimated at approximately 1–7%, although the incidence of paraproteinemia may escalate to 40% (1). A monotypic expression of immunoglobulin light chains and/or B-cell monoclonal proliferation are frequently detected, whereas anti-cytokeratin antibodies stains are constantly negative. The possibility that PLCNA may correspond to a skin-limited localized plasma cell dyscrasia has been hypothesized. Moreover, PLCNA has been documented in association with Sjögren’s syndrome (25%) (3) and CREST syndrome. Upon revisiting the association between PLCNA and CREST syndrome, the reported cases predominantly involved female patients (84%) with a median age of 61.5 years and 80% of the lesions were localized in the lower extremities, presenting a contrast to the facial and acral lesions typically observed in PLCNA cases not associated with CREST syndrome. On average, PLCNA manifested in patients with CREST syndrome after a mean evolution period of 7.9 years; the mean duration of PCLNA until diagnosis was 4.7 years (4–6).
PLCNA lacks a standard treatment, with options like surgical excision (7), shave excisions, and laser therapies aimed at improving lesion appearance. However, recurrence is common, and dyspigmentation, infection, and scarring may occur.
The observation of progressive yellowish, dusky-red papules, plaques, or nodules in a patient with limited systemic sclerosis should prompt consideration of primary cutaneous nodular amyloidosis. Reviewing the literature, a female predilection and a tendency for lower extremity involvement become apparent when considering the PLCNA and CREST syndrome association. The mean time for patients with CREST syndrome to develop PLCNA was approximately 8 years.
REFERENCES
- Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol 2001; 145: 105–109.
- Molina-Ruiz AM, Cerroni L, Kutzner H, Requena L. Cutaneous deposits. Am J Dermatopathol 2014; 36: 1–48.
- Llamas-Molina JM, Velasco-Amador JP, De la Torre-Gomar FJ, Carrero-Castaño A, Ruiz-Villaverde R. Localized cutaneous nodular amyloidosis: a specific cutaneous manifestation of Sjögren’s syndrome. Int J Mol Sci 2023; 24: 73–78.
- Goettsche LS, Moye MS, Tschetter AJ, Stone MS, Wanat KA. Three cases of localized cutaneous nodular amyloidosis in patients with limited systemic sclerosis and a brief literature review. Int J Womens Dermatol 2017; 3: 91–95.
- Atzori L, Ferreli C, Matucci-Cerinic C, Pilloni L, Rongioletti F. Primary localized cutaneous nodular amyloidosis and limited cutaneous systemic sclerosis: additional cases with dermatoscopic and histopathological correlation of amyloid deposition. Dermatopathology 2021; 8: 229–235.
- Kikuchi N, Sakai E, Nishibu A, Ohtsuka M, Yamamoto T. Primary localized cutaneous amyloidosis in patients with scleroderma. Acta Derm Venereol 2010; 90: 326–327.
- Joo KI, Yoo DS, Roh MR. Primary localized cutaneous nodular amyloidosis on scalp successfully treated with excision. Ann Dermatol 2020; 32: 327–330.