REVIEW ARTICLE
Malignant Transformation in Porokeratosis Ptychotropica: A Systematic Review
Chee H. LOH1, Chris L. TAN1, Kong B. TAN2, Holger SUDHOFF3 and Peter GOON1,4
1Department of Dermatology, National University Hospital, Singapore, 2Department of Pathology, National University Hospital, Singapore, 3Department of Otolaryngology, University of Bielefeld, Germany, and 4Department of Medicine, National University of Singapore, Singapore
Porokeratosis ptychotropica (PP) is a rare and unusual variant of porokeratosis. There is a dearth of information on the natural history, epidemiology, and optimal treatment options. This study aimed to characterize the worldwide distribution, epidemiology, clinical features, and treatments attempted for all reported cases of porokeratosis ptychotropica. A total of 59 cases of porokeratosis ptychotropica have been reported, with most cases originating from the United States. The median age of patients affected with porokeratosis ptychotropica was 49 years. The most involved body locations are the buttocks and gluteal cleft. The risk of malignant transformation in porokeratosis ptychotropica is approximately 1.7% but there is significant bias in estimating rare occurrences in rare diseases. In conclusion, PP is an important but under-recognized variant of porokeratosis, with a likely low risk of malignant transformation. The best available treatment modality remains uncertain; however, the use of topical lovastatin/cholesterol cream appears promising. Long-term surveillance appears prudent for porokeratosis ptychotropica due to a risk of cancerization.
Key words: porokeratosis; porokeratosis ptychotropica; genitogluteal porokeratosis
SIGNIFICANCE
Porokeratosis ptychotropica is a rare disease affecting the genitogluteal region. The natural history, epidemiology, and optimal treatment options are not well understood. We confirmed that the reported worldwide frequency of porokeratosis ptychotropica is extremely low. Porokeratosis ptychotropica also appears to be associated with a lower risk of malignant transformation compared with other commoner forms of the disease but there are inherent difficulties in studying the natural history of extremely rare diseases. The most involved body locations were the buttocks and gluteal cleft. The optimal treatment option remains uncertain, but topical lovastatin/cholesterol cream is a promising treatment option. It is important to be cognizant of this under-recognized condition to avoid delayed diagnosis.
Citation: Acta Derm Venereol 2024; 104: adv40558. DOI: https://doi.org/10.2340/actadv.v104.40558.
Copyright: 2024 © The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Apr 17, 2024. Accepted after revision: Aug 6, 2024. Published: Oct 10, 2024
Corr: Chee H. Loh, Department of Dermatology, NUHS Tower Block Level 10, 1E Kent Ridge Road, Singapore 119228. E-mail: cheehoou.loh@mohh.com.sg
Competing interests and funding: The authors have no conflicts of interest to declare.
INTRODUCTION
Porokeratosis is a genodermatosis with clinically distinct phenotypes such as disseminated superficial actinic porokeratosis (DSAP), disseminated superficial porokeratosis (DSP), linear porokeratosis (LP), punctate porokeratosis (PuP), porokeratosis of Mibelli (PM), and porokeratosis ptychotropica (PP). PP is a rare and unusual variant of porokeratosis that involves the genitals and gluteal folds, buttocks, and perianal area.
The PP variant is the most infrequently reported of all the phenotypes, as it involves the perineal and perianal areas, and therefore has a resultant concomitant lack of ultraviolet (UV) exposure in most people. UV exposure stands out as the most important mutagen for skin malignancies. Indeed, a recent study (1) concluded that patients with porokeratoses had a much-increased risk of developing skin malignancies (hazard ratio of 4.3 for squamous cell carcinoma, 2.4 for basal cell carcinoma, and 1.83 for melanoma). Inci et al. (1) reported only 2 cases of the genitogluteal or PP variant out of a cohort of 108 (1.8%) cases collected from 2016 to 2020.
The term “porokeratosis ptychotropica” is derived from the Greek words ptyche (fold) and trope (a turning), referring to the flexural nature of this condition. Affected patients commonly present with pruritic verrucous plaques affecting the buttocks, most commonly over the inter-gluteal regions. Relevant differential diagnoses to consider include psoriasis, verruca vulgaris, chronic intertrigo, lichenified eczema, and lichen planus. There is a dearth of information on the natural history, epidemiology, and treatments for this rare condition.
In this article, we report a rare case of PP in Singapore, which triggered this review due to the scarcity of knowledge on frequency, management, and risk of malignancy. Therefore, we have explored published case reports to ascertain the worldwide geographic frequency of this unusual variant, and more importantly to estimate the risk or likelihood of malignant transformation of these lesions. Finally, we review the success or failure of the multiple different treatment modalities used worldwide.
CASE HISTORY
A 56-year-old man with no significant past medical history presented to our academic medical centre in October 2022, with a 3-year history of a pruritic dermatosis over the gluteal cleft. He responded initially to mometasone cream but later developed worsening rashes over the same area. He is an avid cycler and wears tight cycling shorts in the tropical weather of Singapore. Physical examination revealed brown scaly papules with a hyperkeratotic rim, scattered over both sides of the gluteal cleft (Fig. 1). There was also a scaly lichenified plaque with a violaceous border over the medial aspects of both gluteal folds, and numerous smaller satellite lesions, which were reported to have developed after the primary large lesion.
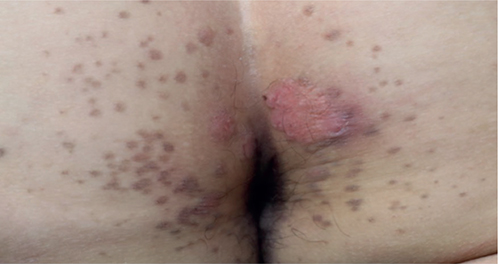
Fig. 1. Verrucous pink plaques over the gluteal folds, surrounded by satellite red-brown papules with a peripheral hyperkeratotic rim.
A punch biopsy was performed, which showed a tiered column of parakeratosis (cornoid lamellae) with mild dermal superficial perivascular infiltrate of lymphocytes (Fig. 2). Correlating the histopathological findings with the clinical presentation, a diagnosis of porokeratosis ptychotropica was established.
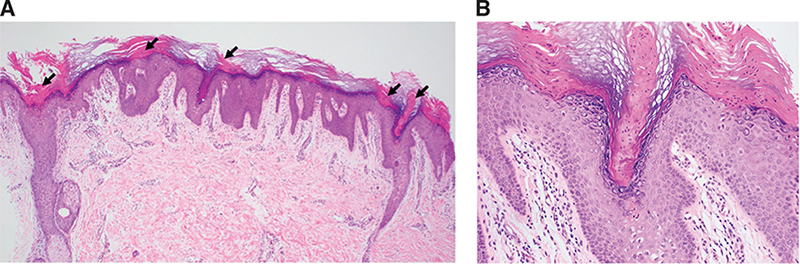
Fig. 2. (A) Low-power view showing epidermal hyperplasia with columns of parakeratosis (arrows) and mild dermal superficial perivascular infiltrate of lymphocytes (haematoxylin & eosin 40x magnification). (B) High-power view showing column of parakeratosis (cornoid lamella) with loss of granular layer and underlying dyskeratotic keratinocytes (haematoxylin & eosin 200x magnification).
MATERIALS AND METHODS
We conducted a comprehensive search for and review of all reported cases in the major databases including PubMed, Scopus, Web of Science, and Google Scholar. All included patients are biopsy-proven cases of PP. Search terms used during the literature search process include “porokeratosis” and “porokeratosis ptychotropica”. Only publications written in the English language were included in this analysis. The year of publication ranged from 1995 to 2022. The selection of articles for inclusion and data extraction was performed independently by CHL and PG; any disagreements were resolved by consensus. A risk-of-bias assessment of all included studies was performed (Table SI). This systematic review was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis guidelines (Fig. S1). This study was registered with the PROSPERO database (CRD42023486488).
To estimate the incidence and prevalence of such a rare entity, we have had to make several assumptions:
(i) The symptomatology of itching, discomfort, and the appearance of these lesions in the anogenital area is likely to drive the patient to seek a medical opinion.
(ii) There is a significant likelihood of case reporting of histologically proven PP due to its inherent rarity.
(iii) There is a bias towards countries with a better developed healthcare system, and dermatologists to undertake biopsies, to report these cases.
(iv) There is likely to be increased surveillance of these patients once diagnosed, due to dermatologist/colorectal surgeon/urological surgeon interest in such a rare condition, plus the reported increased risk of malignant transformation of these lesions.
RESULTS
Over a period of 27 years from 1995 to 2022, a total of 59 cases of PP have been reported in the literature (Table SII). Of the 59 reported cases of PP, the mean age of patients affected with PP was 46.3 years, with a median age of 49 years. The youngest patient affected was 3 years old, and the oldest patient diagnosed was 84 years old. Of these, 51 cases were male (86.4%), and 7 patients were female (11.9%) (Table I). This gives a male:female ratio of approximately 9:1. The most involved body locations are the buttocks, followed by the gluteal cleft. Other less commonly involved locations include scrotal skin, glans penis, and lower extremities. Of the cases reported, characteristic clinical features include well-demarcated red-brown scaly or verrucous plaques with raised keratotic borders, and peripheral lesions. The mean duration of lesions at the time of presentation was 9.8 years.
Despite the inherent biases of studying a rare disease, our analyses revealed the following: the crude cumulative incidence rate in the population of Singapore is likely to be approximately 2/(AUC of Singapore’s population curve)/10 (in a 10-year period), which is equivalent to 21.3 × 10–10 per year. Similar calculations for worldwide incidence rates give us a crude cumulative incidence rate of 2.16 – 4.28 × 10–10 per year. As previously discussed, there is likely to be under-reporting and therefore these figures are likely to be underestimates of the true incidence of disease but also confirm the rarity of reported cases worldwide.
The world heat map of cases (Fig. 3) demonstrates that the majority of reported cases originated from the United States, China, and the rest of the Asia-Pacific region. A disproportionally higher number of cases were reported from the United States (Table II), most likely for the reasons already discussed.
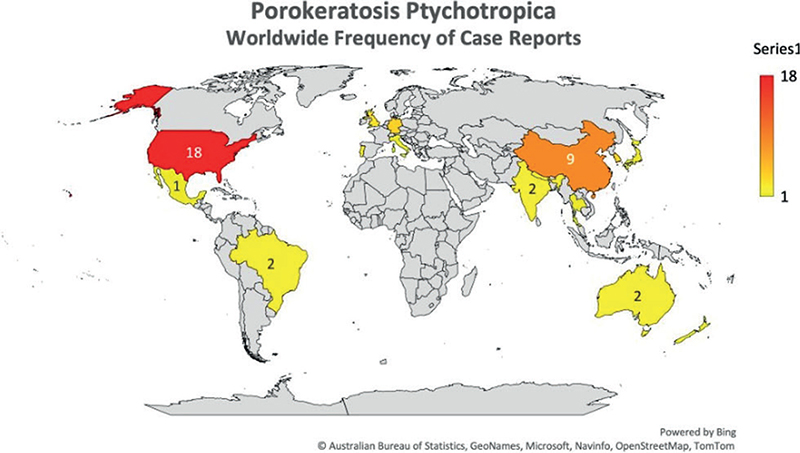
Fig. 3. Frequency heat map of reported cases of porokeratosis ptychotropica.
The rate of malignant transformation of PP is of particular interest. Only 1 case of PP has been reported to undergo malignant transformation (2). Mazori et al. (2) highlighted the fact that transformation into squamous cell carcinoma (in one part of the lesion) occurred over a period of 18 years after initial presentation, and that the only identifiable risk factor was that 30 years previously the patient had suffered cervical carcinoma, which was treated by abdominal hysterectomy and bilateral salpingo-oophorectomy, plus adjuvant Cobalt-60 external beam radiotherapy (ionizing radiation).
There have been a multitude of treatment modalities reported for PP (Table I). Most patients have received either single or combination therapies. In 12 of the reported cases, details on the treatment administered was not provided. Topical retinoids were the most utilized method of treatment (22.0%), followed by oral retinoids (18.6%) and topical corticosteroids (15.2%). Topical or oral retinoids are favoured choices of treatment as porokeratosis is thought to be a disorder related primarily to epidermal keratinization. No treatment modality has been shown to be consistently efficacious for PP, and patients have often experienced varied success with different treatment options (3–5). In this review, 22 out of 59 patients (37.3%) reported unsatisfactory response to treatment, whereas only 14 out of 59 patients (23.7%) reported satisfactory response to treatment. In a retrospective study on skin-of-colour patients, 16% of patients demonstrated good response while 53% of patients experienced a partial response to treatment (6).
Recently, a novel topical treatment targeting the HMG-CoA reductase enzyme (using 2% lovastatin), and replacement of cholesterol as end-product of the cholesterol synthesis pathway (2% cholesterol), has been described in treating other variants of porokeratosis (7). A few small studies and case reports have reported success using this compound topical as treatment (8–10).
DISCUSSION
Lucker et al. (11) reported the first case of PP, which shared similar clinical and histological features to our case. Since then, although there have been infrequent case reports of PP in the literature, little is known about the epidemiology and natural history of the disease. This study confirms that PP is highly infrequent (worldwide crude incidence rate approximated at 2.16–4.28 × 10–10 per year (12) but with the caveats and biases in analyses that we have previously discussed. In comparison, the US incidence rate is higher than that of the worldwide incidence rate. This can be attributed to the increased likelihood of reporting a rare disease in the United States, owing to the availability of advanced healthcare infrastructure and greater access to dermatological care as compared with the rest of the world. Other variants of porokeratosis appear to be much more frequent, particularly DSAP (1).
We observed an overwhelming male predominance in PP, which is consistent with what Das et al. (13) had analysed in their review. This is in contrast with a case series conducted by Hsueh et al., who showed a male predominance in DSP and a female predominance in the DSAP group (14). The reasons for this remain to be determined but may be related to a difference in sex hormone (androgenic as opposed to oestrogenic) involvement, perhaps a reduced sensitivity of male patients to attend their doctors with problems involving their genital region, and, finally, that UV exposure is unlikely to be involved in the pathogenesis of PP (no increased reports of nude sunbathing etc. in the male patients reported).
Malignant transformation of porokeratosis is of singular importance, and the risk of this process is the main reason for continued long-term surveillance of patients. The risk of malignant transformation has been estimated at 7.5% in general for porokeratosis of all types, lower for DSAP (3.4%) but higher (19%) for linear porokeratosis (15). Otsuka et al. (16) reported that 11.6% of their cohort of 198 patients developed SCC, Bowen’s disease ± basal cell carcinoma in their porokeratotic lesions. A more recent single-centre study had attempted to eliminate publication bias by conducting a retrospective 18-year (2000–2017) study retrieved from their institutional databases and concluded that 29.3% of their DSAP patients developed malignancies within their lesions, and 2/2 (100%) of their LP patients developed malignancies within their lesions (17). It is clear that there is considerable difference between estimates in the published literature.
Our own analyses here of all worldwide published PP cases give a crude estimate of the risk of malignant transformation in PP and confirm that it is likely to be low, but this figure is subject to inherent biases of different histological availabilities, dermatological personnel and facilities, competencies, access to medical care, etc. As previously discussed, there is likely to be under-reporting and therefore these calculations are likely underestimates of the true global incidence.
It is important to note that in the 1 reported case of squamous cell carcinoma (SCC) in PP (2), there was a history of ionizing radiation, with no detectable HPV 6, 11, 16, or 18. Other known high-risk HPV subtypes recognized to be important in cervical carcinoma or anal carcinoma were not tested for. Further, a recent Swedish nationwide registry cohort study demonstrated significant associations between porokeratosis and skin cancer, with significant hazard ratios of increased risks for squamous cell carcinoma (4.3, 95% CI of 3.4–5.4), basal cell carcinoma (2.42, 1.97–2.98), and melanoma (1.83, 1.18–2.82) (1). It is therefore most prudent to conclude that patients with PP will require long-term surveillance for malignancy development, despite the uncertainty over the magnitude of risk. Known risk factors for malignancy development in porokeratosis lesions include size of lesion, duration of lesion development, acral involvement, and ionizing radiation (including actinic damage) (15, 16, 18).
HPV is designated a Class 1 carcinogen by the World Health Organization, and therefore the fact that the patient had previously suffered cervical carcinoma (> 97% HPV positive worldwide) years ago meant that it was imperative that HPV status of the new SCC was tested for. Mazori et al. (4) reported that the case of PP transformed into SCC was tested for HPV 6, 11, 16, and 18 by in-situ hybridization (ISH) and this was negative, and the SCC also tested negative for p16 expression but positive for p53 and cyclin D1. The lack of testing for other high-risk HPV subtypes is inadequate as at least 15 HPV subtypes known to be of the high-risk variety for malignant transformation in cervical carcinoma. An HPV-induced SCC in the anogenital region usually tests strongly positive for both p16 and p53 expression. Further, it was reported that the tumour displayed diffuse positivity for cyclin D1. Cyclin D1a isoform has been reported to be increased in cervical tumour tissue compared with CIN (cervical intra-epithelial tissue) but similar to normal tissue. Cyclin D1b isoform has been reported to be expressed at higher levels in normal tissue compared with tumour tissue (19). Since Cyclin D1 isoform was not reported in the paper by Mazori et al. (2), its relevance as a useful biomarker in that case is negligible. Despite that, the report demonstrates that PP lesions can transform into malignant tissue, although the rate of transformation is not high, perhaps only of the order of 1/59 (1.7%) if we presume that all 59 cases we found in literature were followed up. This contrasts somewhat with the risk of malignancy reported for disseminated superficial actinic porokeratosis (DSAP) of 3.4% to 19% in linear porokeratosis (15). These figures contrast with a more recent single-centre study, which reported 29.2% of their DSAP (12/41) and 100% of linear porokeratosis (2/2) transformed (18). It is interesting to speculate that removal of the most dominant risk factor (natural UV light) in the population suffering from PP has resulted in a concomitantly low rate of malignant transformation.
The exact pathophysiology of porokeratosis remains to be determined, but it is thought to be a disorder related to aberrant terminal differentiation of keratinocytes (20). The pathogenesis and development of the porokeratosis phenotype in skin appears to be via a 2-hit process (fulfilling Knudson’s 2-hit hypothesis, although Knudson developed his theory primarily in relation to cancer pathogenesis) (21). The majority of work has been done in the more common variants of porokeratosis such as DSAP and LP (due to the availability of samples), but there is no reason to suppose that the pathway is very different in PP. Familial cases demonstrate transmission via an autosomal dominant modality, with a primary germline mutation in the mevalonate pathway enzymes (usually MVK or MVD), and porokeratotic lesions demonstrate a second hit mutation in the corresponding wild-type allele, allowing development of the classic annular lesion. Sporadic cases develop through a de novo germline mutation in the same pathways at meiosis, and the porokeratotic lesions also demonstrate the second-hit mutation in the wild type allele. Kubo et al. (22) demonstrated that the phenotypic lesion develops clonally from second-hit cells, resulting in an expanding rim of first-hit, second-hit, and infiltrating mononuclear leucocytes. The centre of the lesion was almost entirely composed of second-hit cells, suggesting that it originated from here and expanded outwards, abutting on normal first-hit cells. This model of pathogenesis requires verification and confirmation through detailed molecular analyses of PP tissue.
A variety of modalities have been attempted for the treatment of PP (Table III). In the majority of these cases, combinations of topical, oral, and destructive therapeutic options have been utilized. However, topical and oral options have generally been reported as unsatisfactory, and this is especially pertinent to PP as seen in our review of all the treatments tried so far. The novel targeted topical treatment using 2% Lovastatin to inhibit HMG-CoA reductase, +/- replacement of cholesterol (using 2% cholesterol) as end-product in this cholesterol synthesis pathway, holds some promise as an efficacious treatment, with very few reported adverse effects. We will await studies using this topical treatment in this variant of porokeratosis (PP), as there is no reason to assume that a different molecular pathogenesis in involved in PP.
We conclude here by reiterating that the rarity of PP and the multitude of treatments used in an attempt to treat the lesion successfully demonstrates the uncertainty of best available management. However, a recent novel targeted topical treatment may hold some promise and represents an exciting step forward. In short, we have conducted a review of worldwide reports of PP diagnoses, confirmed that the reported worldwide frequency of this rare variant of porokeratosis is very low, discussed the molecular pathogenesis of this genodermatosis, and approximated the risks of transformation. Surveillance (and treatment if available) appears to be mandatory for PP and other variants of porokeratoses.
REFERENCES
- Inci R, Zagoras T, Kantere D, Holmström P, Gillstedt M, Polesie S, et al. Porokeratosis is one of the most common genodermatoses and is associated with an increased risk of keratinocyte cancer and melanoma. J Eur Acad Dermatol Venereol 2022; 37: 420–427. https://doi.org/10.1111/jdv.18587
- Mazori DR, Shvartsbeyn M, Meehan SA, Tarsis SL. Transformation of porokeratosis ptychotropica into invasive squamous cell carcinoma. Int J Dermatol 2017; 56: 679–680. https://doi.org/10.1111/ijd.13575
- Stone N, Ratnavel R, Wilkinson JD. Bilateral perianal inflammatory verrucous porokeratosis (Porokeratosis ptychotropica). Br J Dermatol 1999; 140: 553–555. https://doi.org/10.1046/j.1365-2133.1999.02738.x
- Pitney L, Weedon D, Pitney M. Porokeratosis ptychotropica: a rare variant with discrete lesions. Australas J Dermatol 2015; 56: e28–29. https://doi.org/10.1111/ajd.12213
- Scheiba N, Enk A, Proske S, Hartschuh W. Porokeratosis ptychotropica: successful treatment with the dermatome. Dermatol Surg 2010; 36: 257–260. https://doi.org/10.1111/j.1524-4725.2009.01402.x
- Tan LS, Chong WS. Porokeratosis in Singapore: an Asian perspective. Australas J Dermatol 2012; 53: e40–44. https://doi.org/10.1111/j.1440-0960.2011.00856.x
- Atzmony L, Lim YH, Hamilton C, Leventhal JS, Wagner A, Paller AS, et al. Topical cholesterol/lovastatin for the treatment of porokeratosis: a pathogenesis-directed therapy. J Am Acad Dermatol 2020; 82: 123–131. https://doi.org/10.1016/j.jaad.2019.08.043
- Santa Lucia G, Snyder A, Lateef A, Drohan A, Gregoski MJ, Barton V, et al. Safety and efficacy of topical lovastatin plus cholesterol cream vs topical lovastatin cream alone for the treatment of disseminated superficial actinic porokeratosis: a randomized clinical trial. JAMA Dermatol 2023; 159: 488–495. https://doi.org/10.1001/jamadermatol.2023.0205
- Byth LA, Byth J. Topical simvastatin-cholesterol for disseminated superficial actinic porokeratosis: an open-label, split-body clinical trial. Australas J Dermatol 2021; 62: 310–313. https://doi.org/10.1111/ajd.13601
- Diep D, Janitz T, Kannan KS, Crane A, Aluri B, Wright K, et al. Bilateral linear porokeratosis treated with topical cholesterol 2%/lovastatin 2. Cureus 2022; 14: e27540. https://doi.org/10.7759/cureus.27540
- Lucker GP, Happle R, Steijlen PM. An unusual case of porokeratosis involving the natal cleft: porokeratosis ptychotropica? Br J Dermatol 1995; 132: 150–151. https://doi.org/10.1111/j.1365-2133.1995.tb08643.x
- Tasker F, Lewis F, Kemp N, Lebas E, Calonje JE. A hidden and rare variant of porokeratosis: a case report of porokeratosis ptychotropica and report of the literature. Australas J Dermatol 2022; 63: 495–496. https://doi.org/10.1111/ajd.13935
- Das A, Vasudevan B, Talwar A. Porokeratosis: an enigma beginning to unravel. Indian J Dermatol Venereol Leprol 2022; 88: 291–299. https://doi.org/10.25259/IJDVL_806_20
- Hsueh Y-T, Hsu T-C, Hsu C-K, Lee JY-Y, Yang C-C. Disseminated superficial porokeratosis and disseminated superficial actinic porokeratosis: a case series of 39 patients. Dermatologica Sinica 2020; 38: 221–224. https://doi.org/10.4103/ds.ds_41_20
- Sasson M, Krain AD. Porokeratosis and cutaneous malignancy: a review. Dermatol Surg 1996; 22: 339–342. https://doi.org/10.1111/j.1524-4725.1996.tb00327.x
- Otsuka F, Someya T, Ishibashi Y. Porokeratosis and malignant skin tumors. J Cancer Res Clin Oncol 1991; 117: 55–60. https://doi.org/10.1007/BF01613197
- Novice T, Nakamura M, Helfrich Y. The malignancy potential of porokeratosis: a single-center retrospective study. Cureus 2021; 13: e13083. https://doi.org/10.7759/cureus.13083
- Maubec E, Duvillard P, Margulis A, Bachollet B, Degois G, Avril MF. Common skin cancers in porokeratosis. Br J Dermatol 2005; 152: 1389–1391. https://doi.org/10.1111/j.1365-2133.2005.06639.x
- Gu J, Zhang X, Yang Z, Wang N. Expression of cyclin D1 protein isoforms and its prognostic significance in cervical cancer. Cancer Manag Res 2019; 11: 9073–9083. https://doi.org/10.2147/CMAR.S224026
- Kamata Y, Maejima H, Watarai A, Saito N, Katsuoka K, Takeda A, et al. Expression of bleomycin hydrolase in keratinization disorders. Arch Dermatol Res 2012; 304: 31–38. https://doi.org/10.1007/s00403-011-1180-6
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer 2001; 1: 157–162. https://doi.org/10.1038/35101031
- Kubo A, Sasaki T, Suzuki H, Shiohama A, Aoki S, Sato S, et al. Clonal expansion of second-hit cells with somatic recombinations or C>T transitions form porokeratosis in MVD or MVK mutant heterozygotes. J Invest Dermatol 2019; 139: 2458–2466.e59. https://doi.org/10.1016/j.jid.2019.05.020