SHORT COMMUNICATION
Anti-p200 Pemphigoid Mimicking Acquired Reactive Perforating Collagenosis
Janik FLEISSNER*, Katrin ICKRATH, Matthias GOEBELER, Sandrine BENOIT and Andreas KERSTAN
Department of Dermatology, Venereology and Allergology, University Hospital Würzburg, Josef-Schneider-Straβe 2, DE-97080 Würzburg, Germany. *E-mail: Fleissner_j@ukw.de
Citation: Acta Derm Venereol 2024; 104: adv40627. DOI: https://doi.org/10.2340/actadv.v104.40627.
Copyright: © 2024 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Apr 19, 2024; Accepted after revision: Aug 12, 2024; Published: Sep 9, 2024
Competing interests and funding: The authors have no conflicts of interest to declare.
This work was supported by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF), University Hospital Würzburg, Germany (AdvCSP-2 to AK) and by the Else-Kröner-Fresenius-Stiftung (TWINSIGHT-CSP to JF).
INTRODUCTION
Anti-p200 pemphigoid, first described in 1996, is a rare autoimmune blistering dermatosis with an annual incidence of 0.7 patients per 1 million inhabitants (1–3). Typically, it presents in younger age than bullous pemphigoid (BP) and demonstrates a male predilection. Notably, over half of Japanese cases are preceded by psoriasis, a feature observed in only about 6% of non-Japanese patients (4). Pathogenetically, autoantibodies elicit dermoepidermal separation, with Laminin β4 recently identified as the primary target antigen, while anti-Laminin γ1 autoantibodies are detectable in 70–90% of cases (5, 6). Clinically, anti-p200 pemphigoid closely mimics BP, marked by intense itching, tense blisters on erythematous skin, and urticarial plaques (7). Predilection sites include the distal extremities, and approximately 40% of cases involve the oral and genital mucosa (4). However, in rare cases, anti-p200 pemphigoid exhibits a clinically diverse spectrum, resembling dermatitis herpetiformis, erythema multiforme, linear IgA bullous dermatosis, and pompholyx (7, 8). This case report presents an atypical case of anti-p200 pemphigoid that mimics acquired reactive perforating collagenosis (ARPC) (9).
CASE REPORT
A 73-year-old woman was referred to our department because of excruciating pruritus with vesiculation followed by skin crusting. Symptoms started approximately 1 year before, prompting an external histological examination, which revealed eczema. Initial management included 5% polidocanol in Unguentum Cordes and mometasone furoate cream. Notably, the patient had a 20-year history of seropositive rheumatoid arthritis, initially managed with methotrexate and leflunomide, and later with leflunomide alone. Her medical history was devoid of other dermatoses, diabetes mellitus, and impaired kidney function.
Physical examination on initial presentation revealed numerous plaques up to 10 cm in diameter with raised erythematous borders and central adherent green-brown keratotic plugs (Fig. 1A–C). They were localized on both the trunk and extremities, with the largest one situated distally on the left lower leg. Individual lesions were predominantly round-oval in shape, but sometimes coalesced to form polycyclic keratinized plaques. On the inner side of the right knee, a solitary serosanguineous blister, about 1 cm in size, was identified (Fig. 1D). Additionally, on the left side of her lower lip, there was a superficial erosion, while the remaining visible mucous membranes appeared normal. Upon resolution of the lesions, numerous hypopigmented atrophic scars remained, particularly on the upper dorsum (Fig. 1C, inset).
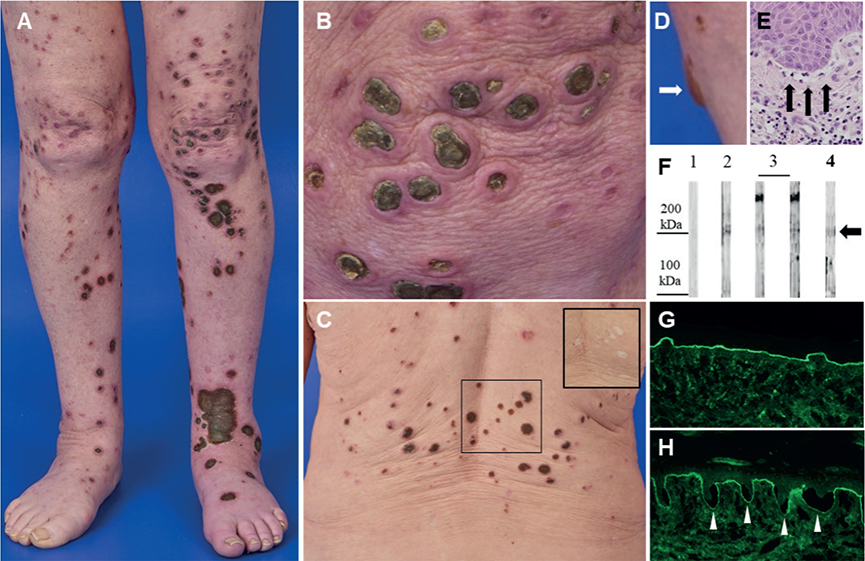
Fig. 1. Clinical features, histopathology, and autoimmune diagnostics. Numerous plaques with raised erythematous borders and central adherent green-brown keratotic plugs on (A, B) the lower limbs and (C) the back. Upon resolution, hypopigmented atrophic scars remain (C, inset). (D) Solitary blister on the inner side of the right knee, indicated by the white arrow. (E) Histopathology of the lesional skin biopsy obtained from the left lower leg exhibiting subepidermal cleft formation as indicated by the black arrows (haematoxylin–eosin [HE], original magnification 20-fold). (F) Human dermis extracts were incubated with serum of a healthy individual (lane 1), control serum of a confirmed anti-p200 patient (lane 2), 2 sera of patients with epidermolysis bullosa acquista (lanes 3), demonstrating a 290 kDa band which represents the alpha chain of type VII collagen, and a 200 kDa band in the serum of our patient (lane 4), indicated by the black arrow. (G) Perilesional direct immunofluorescence of the left lower leg displaying linear basement membrane zone staining with anti-human IgG. (H) Indirect immunofluorescence of human NaCl-split skin stained for IgG (titer 1:320). White arrowheads highlight linear basement membrane zone deposits at the dermal side of the artificial blister.
Given the clinical presentation, we suspected ARPC and hospitalized the patient for comprehensive evaluation and management. The histological analysis of a spindle biopsy obtained from the lesion on the left lower leg revealed the presence of small subepidermal clefts, with no evidence of transepidermal elimination of dermal compounds, as depicted in haematoxylin and eosin (H&E) staining (Fig. 1E) and in sections stained for aldehyde fuchsin to detect elastic fibres (not shown). A mixed inflammatory infiltrate comprising lymphocytes, histiocytes, and neutrophils was observed while eosinophils were not present. However, the differential blood count revealed an eosinophilia of 700/µL (reference: 30–270/µL). Perilesional direct immunofluorescence on the left lower leg demonstrated a linear deposition of IgG (Fig. 1G) and C3, along with a subtle linear deposition of IgA and IgM at the basement membrane. Indirect immunofluorescence on NaCl-separated human split skin displayed the presence of circulating IgG autoantibodies binding to the bottom of the artificial blister at a titre of up to 1:320 (Fig. 1H). Similarly, in indirect immunofluorescence on monkey oesophagus, circulating IgG autoantibodies were detected, exhibiting linear binding to the basement membrane. Immunoblot analyses ruled out the presence of circulating IgG4 autoantibodies against both the immunodominant domain of type VII collagen and Laminin 332. Additionally, indirect immunofluorescence testing for anti-Laminin-332-IgG and an ELISA for anti-collagen-type-VII-IgG both returned negative results. These findings conclusively exclude the differential diagnoses of Laminin 332 pemphigoid and epidermolysis bullosa acquisita. Anti-p200 pemphigoid was confirmed by an external reference laboratory, where circulating IgG1 autoantibodies to the p200 antigen were identified by immunoblotting using human dermis extract (Fig. 1F). Notably, the immunoblot also revealed discrete detection of IgG against Laminin β4.
We treated the patient’s trunk and extremities topically with clobetasol propionate ointment supplemented with 2% triclosan. This facilitated the uncomplicated removal of the soaked crusts. For the underlying erosions, we employed fatty gauze and polyhexanide gel. Given the patient’s significant pain, particularly in the left foot, we administered metamizole and tilidine/naloxone. Additionally, the patient received oral antipruritic therapy with desloratadine 5 mg up to 3 times daily and 5% polidocanol in Unguentum Cordes. We initiated immunomodulatory therapy with doxycycline 100 mg twice daily, which, unfortunately, had to be discontinued after 2 months due to stomach pain and diarrhoea. As disease control with topical treatment alone proved insufficient, we commenced oral systemic therapy with prednisolone at an initial dose of 25 mg/day, equivalent to 0.5 mg/kg bodyweight. After 3 days, we gradually tapered the dose and prematurely discontinued treatment due to the patient’s infection with SARS CoV-2. Nevertheless, there was a notable and stable improvement in the patient’s condition. Aside from hoarseness, the course of SARS-CoV-2 infection remained asymptomatic, with no clinical impact on anti-p200 pemphigoid.
DISCUSSION
In view of our patient’s clinical presentation, our initial suspicion was ARPC. However, the patient’s medical history showed no signs of diabetes mellitus or chronic renal failure. The significant pruritus that led the patient to seek care at our clinic, along with reported blistering preceding plaque formation and histopathological findings of subepidermal clefting, directed our consideration towards autoimmune blistering diseases. Ultimately, anti-p200 pemphigoid was confirmed by an external laboratory.
To date, an association between anti-p200 pemphigoid and histologically proven ARPC has only been documented by Kakehi et al. in a case involving a 71-year-old man with diabetes mellitus who subsequently developed ulcerative colitis (10). Our literature review revealed 10 reported cases linking BP with ARPC. Unlike our case, in these reports (11–14) most patients had diabetes mellitus and were frequently treated with dipeptidyl-peptidase IV (CD26) inhibitors, which are known to promote BP (15). While we could not confirm ARPC histologically in 2 consecutively obtained biopsy specimens, our patient consistently described experiencing blisters followed by erosions and crusting. Similar to Schauer et al., who reported 1 case of BP preceding ARPC-like lesions (13), we suggest that our patient exhibited a rare clinical presentation of anti-p200 pemphigoid. On the other hand, it cannot be ruled out that pre-existing ARPC might have caused tissue damage at the dermoepidermal junction, exposing epitopes that then triggered autoreactive T and B cells to elicit anti-p200 pemphigoid.
Our case underscores the critical need to consider autoimmune blistering dermatoses in patients experiencing severe pruritus, even within atypical clinical presentations. Due to the complexity involved in diagnosing anti-p200 pemphigoid, there is a high likelihood that this rare condition often remains undetected. As such, we emphasize the value of a comprehensive approach that combines detailed clinical evaluations, histological analyses, and autoimmune diagnostics as listed in Table I and recently discussed (6), to obtain precise diagnoses with effective treatments similar to those for BP, such as topical class IV corticosteroids or systemic therapy with prednisolone, dapsone, doxycycline, azathioprine, or mycophenolate (7).
| Clinical features (7) | Diagnostic criteria (4, 6) |
| Blisters on erythematous skin Urticarial plaques Intense pruritus |
Direct immunofluorescence: linear deposition of IgG and/or C3 along the basement membrane zone (BMZ); n-serrated pattern Indirect immunofluorescence: linear BMZ deposition at the dermal side in human NaCl-split skin Immunoblot: p200, Laminin β4, Laminin γ1 Histology: subepidermal cleft |
ACKNOWLEDGMENTS
The authors would like to thank the Laboratory for Cutaneous Autoimmune Diagnostics, led by Prof. Dr. Dr. Enno Schmidt, at the University Hospital Schleswig-Holstein, Campus Lübeck, who performed the p200 and Laminin ß4 immunoblots. This work was supported by the Interdisziplinäres Zentrum für Klinische Forschung (IZKF), University Hospital Würzburg, Germany (AdvCSP-2 to A.K.) and by the Else-Kröner-Fresenius-Stiftung (TWINSIGHT-CSP to J.F.).
REFERENCES
- Chen KR, Shimizu S, Miyakawa S, Ishiko A, Shimizu H, Hashimoto T. Coexistence of psoriasis and an unusual IgG-mediated subepidermal bullous dermatosis: identification of a novel 200-kDa lower lamina lucida target antigen. Br J Dermatol 1996; 134: 340–346. https://doi.org/10.1111/j.1365-2133.1996.tb07625.x
- van Beek N, Weidinger A, Schneider SW, Kleinheinz A, Glaser R, Holtsche MM, et al. Incidence of pemphigoid diseases in Northern Germany in 2016: first data from the Schleswig-Holstein Registry of Autoimmune Bullous Diseases. J Eur Acad Dermatol Venereol 2021; 35: 1197–1202. https://doi.org/10.1111/jdv.17107
- Zillikens D, Kawahara Y, Ishiko A, Shimizu H, Mayer J, Rank CV, et al. A novel subepidermal blistering disease with autoantibodies to a 200-kDa antigen of the basement membrane zone. J Invest Dermatol 1996; 106: 465–470. https://doi.org/10.1111/1523-1747.ep12343631
- Kridin K, Ahmed AR. Anti-p200 pemphigoid: a systematic review. Front Immunol 2019; 10: 2466. https://doi.org/10.3389/fimmu.2019.02466
- Goletz S, Pigors M, Lari TR, Hammers CM, Wang Y, Emtenani S, et al. Laminin beta4 is a constituent of the cutaneous basement membrane zone and additional autoantigen of anti-p200 pemphigoid. J Am Acad Dermatol 2024; 90: 790–797. https://doi.org/10.1016/j.jaad.2023.11.014
- Goletz S, Probst C, Komorowski L, Radzimski C, Mindorf S, Holtsche MM, et al. Sensitive and specific assay for the serological diagnosis of anti-p200 pemphigoid based on the recombinant laminin beta4 subunit. Br J Dermatol 2024; 191: 140–141. https://doi.org/10.1093/bjd/ljae136
- Goletz S, Hashimoto T, Zillikens D, Schmidt E. Anti-p200 pemphigoid. J Am Acad Dermatol 2014; 71: 185–191. https://doi.org/10.1016/j.jaad.2014.02.036
- Semeria L, Lamiaux M, Quinchon JF, Modiano P. Anti-p200 pemphigoid mimicking erythema multiforme. JAAD Case Rep 2022; 21: 157–159. https://doi.org/10.1016/j.jdcr.2022.01.011
- Wagner G, Sachse MM. Acquired reactive perforating dermatosis. J Dtsch Dermatol Ges 2013; 11: 723–729, 723–730. https://doi.org/10.1111/ddg.12131
- Kakehi Y, Miyagawa F, Ogawa K, Hashimoto T, Asada H. Case of anti-laminin gamma1 pemphigoid associated with ulcerative colitis and acquired perforating dermatosis. J Dermatol 2021; 48: e35–e36. https://doi.org/10.1111/1346-8138.15624
- Maki N, Nishie W, Takazawa M, Kakurai M, Yamada T, Umemoto N, et al. Dipeptidyl peptidase-4 inhibitor-associated bullous pemphigoid in a patient with acquired reactive perforating collagenosis. J Dermatol 2018; 45: 600–602. https://doi.org/10.1111/1346-8138.14254
- Nomura H, Mukai M, Niimi Y, Egami S, Yokoyama T, Sugiura M, et al. Coexistence of acquired perforating dermatosis and bullous pemphigoid: three cases. Eur J Dermatol 2017; 27: 192–193. https://doi.org/10.1684/ejd.2016.2944
- Schauer F, Kern JS, Virtic O, Technau-Hafsi K, Meiss F, Thoma K, et al. A new clinical variant of acquired reactive perforating dermatosis-like bullous pemphigoid. Br J Dermatol 2019; 180: 231–232. https://doi.org/10.1111/bjd.17146
- Tani S, Ishii N, Hashimoto T, Tsujioka K. Bullous pemphigoid arising in a patient with acquired perforating dermatosis. Clin Exp Dermatol 2017; 42: 406–409. https://doi.org/10.1111/ced.13080
- Tasanen K, Varpuluoma O, Nishie W. Dipeptidyl peptidase-4 inhibitor-associated bullous pemphigoid. Front Immunol 2019; 10: 1238. https://doi.org/10.3389/fimmu.2019.01238