SHORT COMMUNICATION
Epidermolysis Bullosa Simplex due to a Novel BPAG1-e Homozygous Pathogenic Variant Revealed by Bullous Scabies
Nathalie PIRONON1, Anne WELFRINGER-MORIN2, Stephanie LECLERC-MERCIER3, Emmanuelle BOURRAT2,4 and Alain HOVNANIAN1,5*
1Laboratory of Genetic Skin Diseases, Université Paris Cité, Inserm, UMR 1163, Institut Imagine, 24 Boulevard du Montparnasse, FR-75015 Paris, 2Department of Dermatology, AP-HP, Hôpital Saint-Louis, Paris, 3Department of Pathology, AP-HP, Hôpital Necker-Enfants Malades, Paris, 4Centre de référence maladies rares MAGEC Nord Site Saint-Louis, Hôpital Saint-Louis, Paris, and 5Department of Genomic Medicine of Rare Diseases, AP-HP, Hôpital Necker-Enfants Malades, Paris, France. *E-mail: alain.hovnanian@inserm.fr
Citation: Acta Derm Venereol 2024; 104: adv40691. DOI: https://doi.org/10.2340/actadv.v104.40691.
Copyright: © 2024 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Apr 30, 2024. Accepted after revision: Sep 3, 2024. Published: Dec 12, 2024.
Competing interests and funding: The authors have no conflicts of interest to declare.
INTRODUCTION
Epidermolysis bullosa simplex (EBS) is a heterogeneous group of inherited disorders characterized by skin fragility due to intraepidermal cleavage. Most EBS cases (75%) arise from mutations in KRT5 or KRT14 (1). However, a minority of affected individuals carry mutations in non-keratin genes including dystonin (DST). Alternative splicing of the DST gene, which encodes BPAG1/BP230, leads to several isoforms with different expression levels in the skin, muscles, neurons, and central nervous system (2). Homozygous nonsense variants in the epithelial isoform of BPAG1, BPAG1-e, lead to autosomal recessive EBS with trauma-induced blisters of the skin, fragility of basal keratinocytes, and loss of the inner hemidesmosomal plaques. A dozen cases of DST-associated EBS have been reported to date (3–5). We describe here a patient with very mild skin fragility that was presenting as bullous scabies. Genetic testing identified a previously unreported homozygous frameshift pathogenic variant in the epidermal isoform of DST. The consequences of this variant on the expression of hemidesmosome and focal adhesion molecules was studied by immunostaining.
CASE REPORT
The patient is a 13-year-old Malian boy born from a consanguineous union (Fig. 1A) who was initially referred on suspicion of scabies. Clinical examination revealed profuse tense blisters on the trunk, elbows, and limbs (Fig. 1B, C). Sampling for sarcoptes was highly positive and the patient was put on treatment for bullous scabies. At the follow-up visit, 15 days later, the patient showed marked improvement, but he also reported a history of trauma-induced skin blistering since childhood, which started when he began to walk, and was now confined to the feet and shins after sport (Fig. 1D). He also had palmoplantar keratoderma, tooth enamel dysplasia, and toe nail dystrophy (Fig. 1E, F). No other family member was affected. In view of this chronic bullous dermatosis, a skin biopsy was obtained, which showed a cleavage in the basal epidermal layer (Fig. 1G, H). The dermis under the bulla was not very inflammatory and contained a perivascular inflammatory infiltrate of lymphocytes, histiocytes, and a few neutrophils (data not shown). Transmission electron microscopy showed a normal number of hemidesmosomes (HDs), but their morphology was altered, the HD inner plaque being absent (Fig. 1G, H). Immunofluorescence staining with mAb 279 (Cosmo Bio, Tokyo, Japan) against the C-Terminus of BPAG1-e showed complete absence of BPAG1-e staining in the patient’s skin in contrast to the linear labelling at the dermal–epidermal junction (DEJ) in normal skin (Fig. 2).
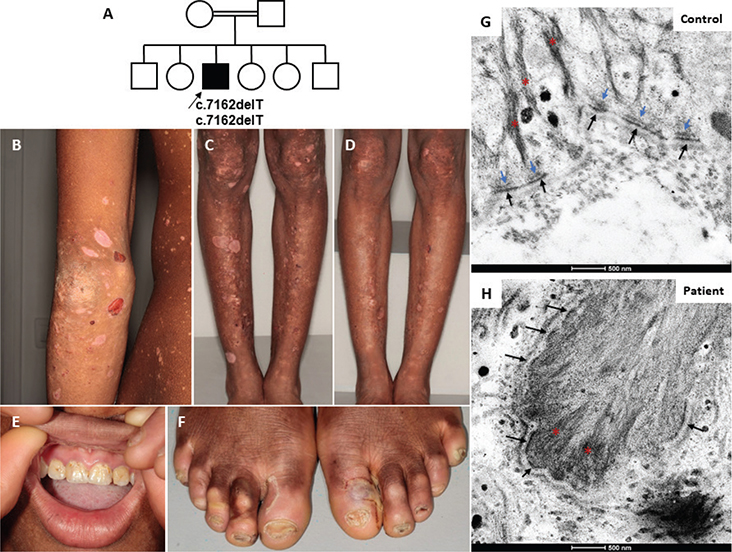
Fig. 1. Clinical phenotype and histopathological findings. (A) Pedigree of the family. Affected index patient (arrow) indicated in black square. (B, C) At the first visit, tense blisters and multiple sequelae of depigmented lesions and rounded hypo-pigmented scars were seen on the patient’s elbows, legs, and knees. (D) 15 days after starting treatment for scabies, slightly dystrophic hypo-pigmented post-bullous scarring was seen. (E) Dental enamel defects of the 2 maxillary incisors. (F) Ungual dystrophy of the 10 toes and a recent blister on the big toe of the left foot. (G, H) Transmission electron microscopy revealed a normal number of hemidesmosomes (HDs) that lack their inner plaque. Blue arrow: HD inner plaque; black arrow: HD outer plaque; red asterisk: keratin intermediate filament. Scale bars = 500 nm.
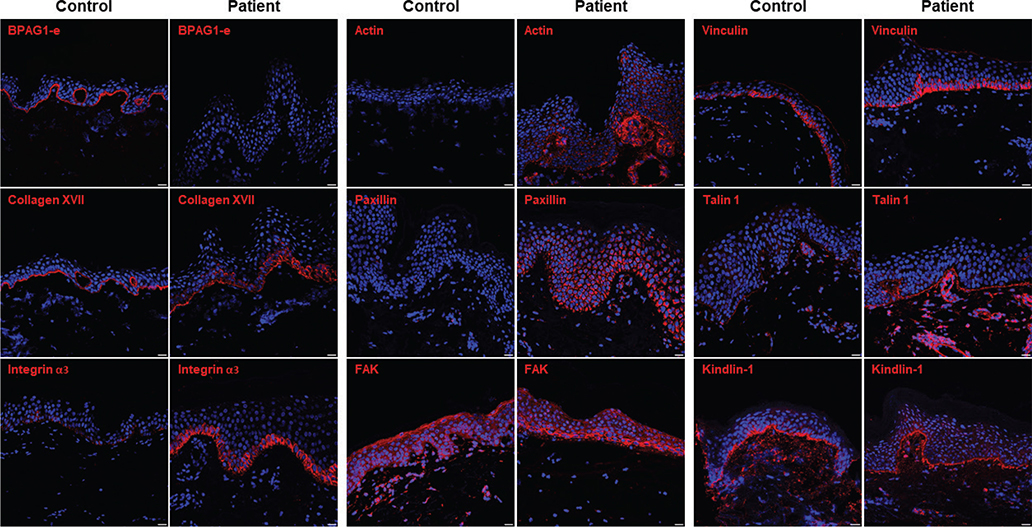
Fig. 2. Immunofluorescence analysis of the patient’s skin. Immunofluorescence staining revealed that BPAG1-e was undetectable in the patient’s skin (Patient) in contrast to linear labelling at the DJE in normal skin (Control). Collagen XVII staining was altered compared with linear deposition along the DEJ in the control skin. FAs proteins (integrin α3, actin, paxillin, focal adhesion kinase [FAK], vinculin, and talin 1) showed increased expression compared with the control. Scale bars = 20 μm. (The settings for image acquisition were made on the image with the strongest signal. Therefore, the strong intensity of the signal in the proband skin prevented visualization of the signal in healthy control skin, which was drastically weaker for several of these FA molecules.)
Next-generation sequencing gene panel analysis identified a homozygous deletion in exon 24, c.7162delT, leading to a frameshift and a premature termination codon (PTC), p.Ser2388Leufs*5 (GenBank NM_001723.6; https://www.ncbi.nlm.nih.gov/genbank/). The mutation reported in our patient is located in the intermediate filament binding domain (IFBD) and is predicted to disrupt the ability of BPAG1-e to bind keratin intermediate filaments. To date, only a few pathogenic DST variants have been reported in the epidermal isoform and not the muscle and nerve isoforms (Fig. S1).
We examined the consequences of this pathogenic variant on the expression of HD and focal adhesion (FAs) proteins by immunostaining. Type XVII collagen (C17) immunolabelling revealed intracellular staining in the patient’s basal cells in contrast to normal healthy subject skin where C17 deposition was linear at the DEJ (Fig. 2 and Fig. S3). Staining for keratin 15 and CD151 was decreased in the patient compared with the control. The pattern and intensity of staining of other HD components (plectin, integrin β4), intermediate filament molecules (keratin 14, keratin 1), and other junctional proteins (laminin α3, laminin γ2) were similar in the patient and control samples (Figs S2 and S3). In contrast, staining intensities of most FAs components (integrin α3, actin, paxillin, focal adhesion kinase (FAK), vinculin, and talin 1) were increased in the patient compared with the control (Fig. 2 and Fig. S3).
DISCUSSION
In this report, bullous scabies revealed a very rare form of EBS resulting from a DST pathogenic variant. It should be noted that bullous scabies revealing or complicating bullous pemphigoid, which results from autoantibodies to BPAG1/BP230, has also been reported. This study extends the spectrum of known DST variants underlying autosomal recessive EB. Compared with other forms of autosomal recessive EB simplex due to KRT14, KRT5, PLEC, EXPH5, and CD151 variants (6), the severity of skin blistering in our patient was relatively mild, as reported in other patients harbouring DST mutations. The homozygous mutation we report led to loss of BPAG1-e protein expression at the DEJ as shown by immunostaining. Considering the terminal location of the variant in the last exon (c.7162delT, p.Ser2388Leufs*5), we expect that nonsense-mediated mRNA decay is not activated. This could lead to the synthesis of a C-Terminus truncated BPAG1-e protein, which would not be detected by the antibody that we used as it specifically recognizes the C-terminal part of the protein. This pathogenic variant is located in a region of the BPAG1-e isoform that is absent from the other tissue isoforms of DST. Immunostaining of other hemidesmosomal components showed abnormal staining of C17 as seen in most cases of EBS resulting from DST variants. This pattern can be explained by decreased incorporation of C17 into the membrane and accumulation in the cells, as suggested by He and colleagues (7). Keratin 15 expression was decreased in our patient, which may be associated with cell stretching, flattening, and migration according to Porter (8). Plectin and β4 integrin subunit staining appeared normal in our patient whereas they were reduced in other EBS-DST cases (Fig. S2). In contrast, the expression of several FAs proteins was increased in the patient’s skin as previously reported in EBS-DST (7).
Cell attachment to the extracellular matrix (ECM) is essential for the integrity and function of multiple tissues. Epithelial cell adhesion is mediated by intermediate filament-associated HDs and actin cytoskeleton-linked FAs. Current evidence suggests that FA dynamics regulate mechanotransduction and cell locomotion (9), while HDs function as stable anchoring adhesion and a prerequisite for apicobasal polarization (10). Using high-resolution imaging, Pora et al. (11) described highly ordered, interdigitated arrays of chevron patterns of HDs and FAs in primary human keratinocytes that seem to depend on each other to sustain directed collective migration.
Wang et al. (12) showed that keratinocytes lacking hemidesmosomal integrin α6β4 exhibit increased FAs formation, cell spreading, and traction-force generation. They demonstrated that impaired HDs assembly leads to increased cellular tension and traction forces and subsequently promotes the maturation of FAs. Additionally, they observed that an increased tension leads to redistribution of integrin αVβ5 from clathrin lattices to FAs. Loss of α6β4 integrin-mediated HDs has also been shown to promote cell migration by stimulating FA dynamics in prostate epithelial cells (13). In our patient, increased expression of FA proteins together with reduced expression of keratin 15 suggest an enhanced state of cell migration.
In summary, we report a patient who presented with bullous scabies but was, after treatment, diagnosed with EBS caused by a novel homozygous DST variant that truncates the C-Terminus of BPAG1-e. We show that loss of function of BPAG1-e increases the expression of FA components, suggesting reorganization of cell-ECM interactions.
ACKNOWLEDGEMENTS
The authors are grateful to Debra-France for their support. They would like to thank the SFR Necker Genomic and Bio-informatic facilities for their help in the NGS sequencing and the SFR Necker imaging facility for its assistance in microscopy imaging.
REFERENCES
- Bolling MC, Lemmink HH, Jansen GHL, Jonkman MF. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol 2011: 637–644. https://doi.org/10.1111/j.1365-2133.2010.10146.x
- Leung CL, Zheng M, Prater SM, Liem RKH. The BPAG1 locus: alternative splicing produces multiple isoforms with distinct cytoskeletal linker domains, including predominant isoforms in neurons and muscles. J Cell Biol 2001; 154: 691–698. https://doi.org/10.1083/jcb.200012098
- Ganani D, Malovitski K, Sarig O, Gat A, Sprecher E, Samuelov L. Epidermolysis bullosa simplex due to bi-allelic DST mutations: case series and review of the literature. Pediatr Dermatol 2021; 38: 436–441. https://doi.org/10.1111/pde.14477
- Wen D, Balacco DL, Bardhan A, Harper N, Walsh D, Ryan G, et al. Localized autosomal recessive epidermolysis bullosa simplex arising from a novel homozygous frameshift mutation in DST (BPAG1). Clin Experimental Derm 2022; 47: 497–502. https://doi.org/10.1111/ced.14917
- Turcan I, Pasmooij AMG, Gostyński A, van den Akker PC, Lemmink HH, Diercks GFH, et al. Epidermolysis bullosa simplex caused by distal truncation of BPAG1-e: an intermediate generalized phenotype with prurigo papules. J Invest Dermatol 2017; 137: 2227–2230. https://doi.org/10.1016/j.jid.2017.04.041
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, Fine J-D, Harper N, Has C, et al. Epidermolysis bullosa. Nat Rev Dis Primers 2020; 6: 78. https://doi.org/10.1038/s41572-020-0210-0
- He Y, Leppert J, Steinke H, Has C. Homozygous nonsense mutation and additional deletion of an amino acid in BPAG1e causing mild localized epidermolysis bullosa simplex. Acta Derm Venereol 2017; 97: 657–659. https://doi.org/10.2340/00015555-2618
- Porter RM, Lunny DP, Ogden PH, Morley SM, McLean WHI, Evans A, et al. K15 expression implies lateral differentiation within stratified epithelial basal cells. Lab Invest 2000; 80: 1701–1710. https://doi.org/10.1038/labinvest.3780180
- De Pascalis C, Etienne-Manneville S. Single and collective cell migration: the mechanics of adhesions. MBoC 2017; 28: 1833–1846. https://doi.org/10.1091/mbc.e17-03-0134
- te Molder L, de Pereda JM, Sonnenberg A. Regulation of hemidesmosome dynamics and cell signaling by integrin α6β4. J Cell Sci 2021; 134: jcs259004. https://doi.org/10.1242/jcs.259004
- Pora A, Yoon S, Windoffer R, Leube RE. Hemidesmosomes and focal adhesions treadmill as separate but linked entities during keratinocyte migration. J Invest Dermatol 2019; 139: 1876-1888.e4. https://doi.org/10.1016/j.jid.2019.03.1139
- Wang W, Zuidema A, te Molder L, Nahidiazar L, Hoekman L, Schmidt T, et al. Hemidesmosomes modulate force generation via focal adhesions. J Cell Biol 2020; 219: e201904137. https://doi.org/10.1083/jcb.201904137
- Schmidt A, Kaakinen M, Wenta T, Manninen A. Loss of α6β4 integrin-mediated hemidesmosomes promotes prostate epithelial cell migration by stimulating focal adhesion dynamics. Front Cell Dev Biol 2022; 10: 886569. https://doi.org/10.3389/fcell.2022.886569