SHORT COMMUNICATION
Nonsense Variant in CYP4F22 Causes Malformation of Corneocyte Lipid Envelopes in a Lamellar Ichthyosis Patient
Ryo FUKAURA, Kana TANAHASHI, Michiya OMI, Takuya TAKEICHI and Masashi AKIYAMA*
Nagoya University Graduate School of Medicine, Department of Dermatology, 65 Tsurumai-cho, Showa-ku, Nagoya-shi, Aichi-ken 466-8560, Japan. *E-mail: makiyama@med.nagoya-u.ac.jp
Citation: Acta Derm Venereol 2025; 105: adv41072. DOI: https://doi.org/10.2340/actadv.v105.41072.
Copyright: © 2025 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Jun 26, 2024. Accepted after revision: Dec 12, 2024. Published: Feb 5, 2025.
Competing interests and funding: The authors have no conflicts to declare.
This research was supported by a Health and Labor Sciences Research Grant for Research on Intractable Diseases (23FC1039) from the Ministry of Health, Labor and Welfare of Japan to M.A. This work was supported by JST FOREST Program grant number JPMJFR210R to TT, by JSPS KAKENHI Grant Number 24K02470 to MA, and by AMED Grant Number JP23ek0109678 to TT and MA.
INTRODUCTION
Cytochrome P450 4F22 (CYP4F22), encoded by CYP4F22, is a protein proposed to be integral to the healthy formation of the corneocyte lipid envelope (CLE) (1). CYP4F22 deficiency clinically results in lamellar ichthyosis (LI), a type of autosomal recessive congenital ichthyosis (ARCI) (1). A number of variants in CYP4F22 have been identified as being pathogenic for LI, with the majority of reports originating from families with Mediterranean or Middle-Eastern backgrounds (2).
Here, we report a patient with LI carrying a novel CYP4F22 variant. In-depth analysis successfully demonstrated the loss of CLE in the stratum corneum, which has not been reported previously.
CASE PRESENTATION
The patient, a 14-year-old girl originally from Afghanistan, presented with erythematous, dry, scaly skin since birth. On examination, there was scaling over almost the entire body, with mild pain and itching (Fig. 1A, B). She was born to consanguineous parents (cousins). Two of her 9 uncles were reported to have similar symptoms. Neither she nor her affected uncles had received any treatment previously.
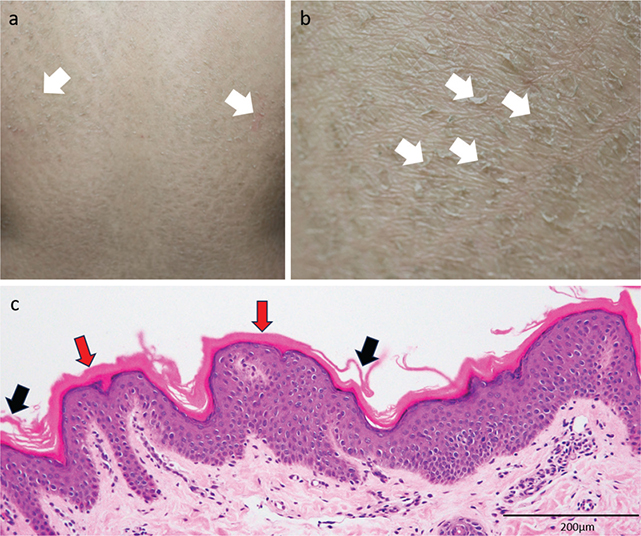
Fig. 1. Clinical and histopathological features of the patient. (A) Photograph of the back of the patient. Some erythema and significant amounts of scaling are seen (white arrows). Similar symptoms were seen over almost the entire body. (B) Photograph of the lower back of the patient. Significant amounts of scaling are seen. Each scale is approximately 1–2 cm in diameter, and the sizes are consistent throughout the back (white arrows). (C) Histopathological photograph of a skin biopsy sample from the right lower back (haematoxylin–eosin staining, magnification x 200). Compact hyperkeratosis can be seen in the majority of the stratum corneum (red arrows), with areas of detachment of the piled stratum corneum cells (black arrows). The epidermis is not thickened, and the stratum granulosum has an unremarkable appearance.
She was started on treatment with emollients and topical steroids. The treatment improved her erythema and scaling to some extent, although she never achieved full remission. She has remained on regular follow-up at our hospital since.
MATERIALS, METHODS AND RESULTS
This study was reviewed and approved by the institutional ethics committee. Informed consent was obtained from the patient and her parents, and all research was performed in accordance with the Declaration of Helsinki.
Genetic analysis was performed using DNA extracted from peripheral blood samples of the patient and her parents. Whole-exome sequencing (WES) was performed to identify pathogenic variants to explain the clinical symptoms, using previously described methods and equipment (2). Variants were filtered according to the characteristics of an autosomal recessive inheritance pattern, and variants with an allele frequency of over 1% in all populations were excluded. Once the candidate variant was identified through WES, Sanger sequencing was undertaken. The region of interest was amplified by polymerase-chain reaction (PCR) using forward (5′-AGCCAACTGCCTGAAATCAT-3′) and reverse (5′-TCAAATGACCCTTCCTCTGG-3′) primers for CYP4F22 exon 4. AmpliTaq Gold 360 DNA Polymerase (Thermo Fischer Scientific. Waltham, MA, USA) was used in the PCR reaction. The resulting PCR products were purified using illustra ExoProStar (Cytiva, Marlborough, MA, USA). Sequencing analysis was performed using the computer software CLC Main Workbench (QIAGEN, Hilden, Germany).
Transmission electron microscopy (TEM) was performed using skin biopsy samples from the patient. The samples were fixed in 2.5% (w/v) glutaraldehyde solution, post-fixed in 0.5% (w/v) ruthenium tetroxide (RuO4) (3), then fixed in an osmium tetroxide solution for 3 h. Subsequent dehydration was performed using ethanol (samples immersed once for 10 min each at concentrations of 50%, 70%, 80%, 90%, 95%, 99%, and 3 times at 100%), before being further immersed in propylene oxide for resin embedding. Once embedding was complete, sections were cut to a thickness of 80 nm using an ultramicrotome UC7k (Leica Microsystems, Wetzlar, Germany) and placed on copper grids. The sections on the copper grids were observed and photographed using an electron microscope JEM-1400PLUS (JEOL, Akishima, Tokyo, Japan).
Immunohistochemical analysis was performed on skin biopsy samples from the patient. The antibodies used were anti-IL-17C, anti-IL-36ɤ, and anti-TNFα using techniques described previously (2). The resulting images were observed and qualitatively assessed. Quantitative analysis was performed using CellProfiler (Broad Institute, Cambridge, MA, USA), software designed to assist image analyses of biological samples. The images were first resized, and redundant areas were crudely cropped and mounted onto the software. A freely available pipeline validated for IHC analysis was used for the software commands (4). The outcomes of interest were the area of visible 3,3′-diaminobenzidine (DAB) staining, the total area of the image, and the stained area as a percentage of the total area (i.e., the area of visible 3,3′-diaminobenzidine staining/total area of the image). This procedure was repeated 3 times, and the average percentage of stained area for each antibody was calculated.
Histopathological analysis of lesional skin biopsied from the right lower back demonstrated an unremarkable appearance of the epidermis. The stratum corneum showed compact hyperkeratosis with less basket-weave appearance (Fig. 1C).
WES identified a homozygous nonsense variant, c.296G > A (p.Trp99Ter), in CYP4F22 in the patient, and the monoallelic identical variant in both of her parents. Given the clinical presentation and the identified variant, the patient was diagnosed with LI.
TEM demonstrated an absence of the CLE around the corneocytes (Fig. S1A, B). Notably, at lower magnification, areas of loosening and scaling could be observed between the layers of corneocytes, suggesting easy skin shedding. Inclusion bodies were also seen in the corneocyte cytosol. In comparison, TEM images of the stratum corneum of a healthy control demonstrated the presence of CLE around the corneocytes (Fig. S1D). All layers of the epidermis except the stratum corneum were found to be intact, and no areas of loosening, scaling, or inclusion bodies were seen (Fig. S1C).
Immunohistochemical analysis with anti-IL-17C, anti-IL-36ɤ, and anti-TNFα antibodies demonstrated an inflammatory response in the patient’s skin compared with controls (Fig. S2). Quantitative analysis using CellProfiler found the area of DAB-stained regions (in pixels) for each antibody to be greater in the patient’s skin (IL-17C 294406px, IL-36ɤ 83109px, TNFα 122562px) than in the controls (IL-17C 69358px, IL-36ɤ 1158px, TNFα 4135px). Furthermore, the average percentage across the 3 measurements for the DAB-stained area was higher for each antibody in the patient’s skin (IL-17C 57.9%, IL-36ɤ 18.3%, TNFα 28.7%) than in the controls (IL-17C 14.7%, IL-36ɤ 0.2%, TNFα 0.8%). A paired t-test showed a statistically significant difference in the percentage of DAB-stained areas for all antibodies (p = 0.01).
DISCUSSION
Patients with LI due to CYP4F22 variants have been reported to possess low levels of acylceramide, required for normal CLE formation (1, 5). In our case, the identified variant in CYP4F22 was thought to lead to absent or reduced CYP4F22 activity, and we hypothesize that less acylceramide would be synthesized, which would explain the absence of CLE. This may be because a premature stop codon appears relatively early in the sequence, which would most likely result in mRNA decay. Even if nonsense-mediated decay did not occur and a truncated protein terminating at the amino acid position 99 were produced, this would still lack a significant proportion of the functional domains of CYP4F22, including the substrate-binding regions (amino acid positions 149–160 and 469–480) (6), and hence would likely be rendered non-functional.
Our preparation of the samples for TEM observation did not use pyridine extraction. Pyridine extraction is used in TEM preparation to enhance the appearance of the CLE and to visualize covalently bound CLE (7). Unbound CLE can be observed without pyridine treatment, which may be the case here. The unbound CLE consists of ultralong-chain acylceramides, cholesterol, and free fatty acids. Of these, the ultralong-chain acylceramides do not have a specific ultrastructure (1, 7). Therefore, even if unbound CLE (mainly free ultralong-chain acylceramides) remained, we think that it is still possible to visualize CLE.
In milder forms of ARCI, such as self-healing collodion baby (SHCB), some production of acylceramide remains, which may contribute to the spontaneous improvement of the ichthyotic phenotype of SHCB with age (2). In contrast, we postulate that a complete lack of CLE, as demonstrated in our case, leads to a chronic phenotype like LI. Indeed, after 2 years of follow-up since the initial presentation, the ichthyotic symptoms persist.
The p.Trp99Ter variant was a nonsense variant that was submitted to ClinVar by 3billion, a South Korean for-profit organization that provides genetic testing services. This particular submission does not mention the clinical details in which it was found (8). It cites the evidence of pathogenicity based on the American College of Medical Genetics guidelines 2015 (9). Therefore, we believe that our description of the present patient and her family is the first published clinical report of this variant, providing further evidence attesting to its pathogenicity and the correlation between mutation severity and phenotype.
The IL-17 family, IL-36 family, and TNFα cytokines have been found to be upregulated in various ARCIs, including Netherton syndrome (10), NIPAL4-associated ichthyosis (11), and SHCB (2). Paller et al. (12) reported significant upregulation of IL-17C and IL-36ɤ in LI patients, although TNFα did not show a significant difference. In our case, TNFα was also significantly increased in lesional skin. The synergistic effect between TNFα and IL-17C has been implicated in various inflammatory conditions (13, 14). The TNFα signalling pathways (e.g., NF-κB, MAPK) are associated with significant tissue inflammation (14, 15), and we feel this may have contributed to the ichthyosis phenotype and may also explain why TNFα was upregulated, unlike previous studies.
In summary, we reported a case of LI arising due to a nonsense variant in CYP4F22. This is the first report in which, through TEM evidence, absent or reduced activity of CYP4F22 has been confirmed to be associated with an absence of CLEs in the stratum corneum. Furthermore, we have confirmed through immunohistochemistry the significant upregulation of pro-inflammatory cytokines associated with ichthyoses as a result of the CYP4F22 variant. We feel that our case provides further clinical evidence of the pathogenesis of LI due to CYP4F22 variants. Analysis of CLEs and other ultrastructural features by TEM might be useful in ARCI diagnosis and in the prediction of long-term prognosis.
ACKNOWLEDGEMENTS
The authors would like to thank Dr Tomoo Ogi for his help in performing whole-exome sequencing, Ms Yuka Terashita for her technical help with preparing samples for TEM observation, and the Division of Medical Research Engineering, Nagoya University Graduate School of Medicine, for allowing them the use of their electron microscope.
REFERENCES
- Akiyama M. Corneocyte lipid envelope (CLE), the key structure for skin barrier function and ichthyosis pathogenesis. J Dermatol Sci 2017; 88: 3–9. https://doi.org/10.1016/j.jdermsci.2017.06.002
- Takeichi T, Ohno Y, Tanahashi K, Ito Y, Shiraishi K, Utsunomiya R, et al. Ceramide analysis in combination with genetic testing may provide a precise diagnosis for self-healing collodion babies. J Lipid Res 2022; 63: 100308. https://doi.org/10.1016/j.jlr.2022.100308
- Crumrine D, Khnykin D, Krieg P, Man M, Celli A, Mauro T, et al. Mutations in recessive congenital ichthyoses illuminate the origin and functions of the corneocyte lipid envelope. J Invest Dermatol 2019; 139: 760–768. https://doi.org/10.1016/j.jid.2018.11.005
- Tollemar V, Tudzarovski N, Boberg E, Törnqvist Andrén A, Al-Adili A, Le Blanc K, et al. Quantitative chromogenic immunohistochemical image analysis in CellProfiler Software. Cytometry 2018; 93: 1051–1059. https://doi.org/10.1002/cyto.a.23575
- Ohno Y, Nakamichi S, Ohkuni A, Kamiyama N, Naoe A, Tsujimura H, et al. Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc Natl Acad Sci U S A 2015; 112: 7707–7712. https://doi.org/10.1073/pnas.1503491112
- Hotz A, Bourrat E, Küsel J, Oji V, Alter S, Hake L, et al. Mutation update for CYP4F22 variants associated with autosomal recessive congenital ichthyosis. Hum Mutat 2018; 39: 1305–1313. https://doi.org/10.1002/humu.23594
- Meyer J, Crumrine D, Schneider H, Dick A, Schmuth M, Gruber R, et al. Unbound corneocyte lipid envelopes in 12R-lipoxygenase deficiency support a specific role in lipid-protein cross-linking. Am J Path 2021; 191: 921–929. https://doi.org/10.1016/j.ajpath.2021.02.005
- National Library of Medicine. NM_173483.4(CYP4F22):c.296G>A (p.Trp99Ter) AND Autosomal recessive congenital ichthyosis 5. Available from: https://www.ncbi.nlm.nih.gov/clinvar/RCV002052170.1/ [Accessed 28/02/2024]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. https://doi.org/10.1038/gim.2015.30
- Barbieux C, Bonnet des Claustres M, Fahrner M, Petrova E, Tsoi LC, Gouin O, et al. Netherton syndrome subtypes share IL-17/IL-36 signature with distinct IFN-α and allergic responses. J Allergy Clin Immunol 2022; 149: 1358–1372. https://doi.org/10.1016/j.jaci.2021.08.024
- Murase Y, Takeichi T, Kawamoto A, Tanahashi K, Okuno Y, Takama H, et al. Reduced stratum corneum acylceramides in autosomal recessive congenital ichthyosis with a NIPAL4 mutation. J Dermatol Sci 2020; 97: 50–56. https://doi.org/10.1016/j.jdermsci.2019.12.001
- Paller AS, Renert-Yuval Y, Suprun M, Esaki H, Oliva M, Huynh TN, et al. An IL-17-dominant immune profile is shared across the major orphan forms of ichthyosis. J Allergy Clin Immunol 2017; 139: 152–165. https://doi.org/10.1016/j.jaci.2016.07.019
- Johnston A, Fritz Y, Dawes S, Diaconu D, Al-Attar P, Guzman A, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol 2013; 190: 2252–2262. https://doi.org/10.4049/jimmunol.1201505
- Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno N, Takahashi K, et al. IL-17B and IL-17C are associated with TNF-α production and contribute to the exacerbation of inflammatory arthritis. J Immunol 2007; 179: 7128–7136. https://doi.org/10.4049/jimmunol.179.10.7128
- Nies J, Panzer U. IL-17C/IL-17RE: Emergence of a unique axis in TH17 biology. Front Immunol 2020; 11: 341. https://doi.org/10.3389/fimmu.2020.00341