SHORT COMMUNICATION
Acquired Erythropoietic Protoporphyria Secondary to Myelodysplastic Syndrome: From Mythology to Oncology
Mirjana UROSEVIC-MAIWALD1*, Jivko KAMARACHEV2, Christoph RENNER3 and Anna-Elisabeth MINDER4,5
1Hautärzte-Zentrum am Zürisee, Seefeldstrasse 214, CH-8008 Zürich, 2Department of Dermatology, University Hospital Zurich, 3Klinik für Hämatologie und Onkologie Hirslanden Zürich, 4Division of Endocrinology, Diabetes, Porphyria, Stadtspital Zürich, Triemli, and 5Swiss Reference Center for Porphyrias, Zürich, Switzerland. *E-mail: maiwald@hautaerzte-zz.ch
Citation: Acta Derm Venereol 2024; 104: adv41146. DOI: https://doi.org/10.2340/actadv.v104.41146.
Copyright: © 2024 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Jul 8, 2024. Accepted: Aug 14, 2024. Published: Aug 28, 2024
INTRODUCTION
Herein we report a case of a 73-year-old man with myelodysplastic syndrome (MDS) who developed severe photosensitivity and crusting skin lesions, diagnosed as acquired erythropoietic protoporphyria (EPP). This case highlights the rare association between MDS and acquired EPP, emphasizing the importance of considering haematological disorders in patients with new-onset photosensitivity and the effectiveness of conservative management strategies.
CASE REPORT
A 73-year-old man presented to our dermatology clinic with a history of sun sensitivity and development of crusting skin lesions for the past 2 years. Typically, after 30 min of direct sun exposure, such as while tending to plants in the garden, he would start to feel burning and excruciating pain in the skin on the face, hands, and even fingertips. He was repeatedly forced to go inside to relieve the skin burning. He also noticed crusting skin lesions appearing on the dorsa of the hands within days after sun exposure. As a young man, he reports having spent a lot of time outside and in the sun, without ever having any of the above-described symptoms. His medical history revealed a myelodysplastic syndrome with multilineage dysplasia (MDS-MLD) diagnosed 6 years ago. MDS-MLD did not require any treatment at that time. His medication included fluticasone-salmeterol spray (asthma), atorvastatin (dyslipidaemia), tamsulosin (prostate hyperplasia), and allopurinol (gout).
Clinical examination revealed thickened, lichenified skin with one fresh blister, numerous crusts, indented scars, and milia formation on the dorsa of the hands and some on the back of the neck and face (Fig. 1A). A working diagnosis of photosensitive dermatosis was made, with differential diagnoses encompassing cutaneous porphyrias, photosensitive drug eruption, lupus erythematosus, and phytophotodermatitis. CBC showed anaemia, and reduced thrombocyte and white blood cell count consistent with his MDS. Liver and renal function parameters were normal. There were no detectable autoantibodies associated with connective tissue diseases. Skin biopsy demonstrated subepidermal blistering with scarce inflammatory reaction, suggesting a porphyria (Fig. 1B). A plasma fluorescence scan was positive and revealed a massive increase in total porphyrins at 33.9 umol/L (normal < 1.30) and free protoporphyrin 30 umol/L (normal < 0.20). A diagnosis of erythropoietic protoporphyria (EPP) secondary to MDS was made.
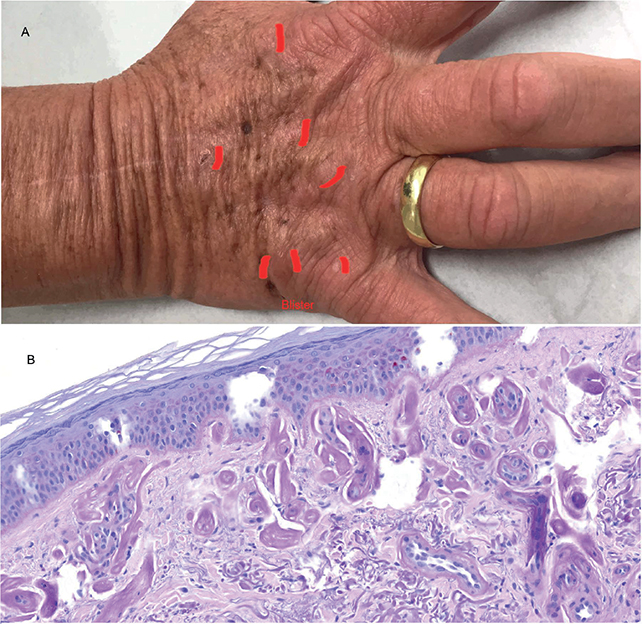
Fig. 1. Skin lesions in acquired erythropoietic protoporphyria. (A) Hand dorsum with scars, crusts, and formation of a blister (all marked in red). (B) Histology of the skin lesions with formation of small subepidermal blisters and thickening of the basal membrane (visualized with PAS-stain, x20 magnification).
DISCUSSION
Porphyrias are a group of rare, typically inherited, metabolic disorders resulting from a deficiency of enzymes involved in heme biosynthesis. The name is derived from the ancient Greek word “porphura”, meaning purple, referring to the purple-pink fluorescence of the teeth and urine seen in some of 8 known porphyria types. EPP is attributed to a deficiency of ferrochelatase (FECH), an enzyme crucial for the final step of heme biosynthesis. This deficiency leads to the accumulation of the metabolite protoporphyrin IX in various tissues (1, 2). In the skin, protoporphyrin IX induces severe photosensitivity through oxidative damage to the skin followed by inflammation (3). Chronic accumulation of protoporphyrin IX in the liver and biliary system can lead to cirrhosis and liver failure (2).
Commonly, EPP follows a semi-dominant mode of inheritance and presents in early childhood upon exposure to sunlight, with symptoms often intensifying in spring and summer. Affected individuals experience intense burning, tingling, or itching on sun-exposed areas of the face and dorsal hand, followed by redness, swelling, and even blister formation. Children with these symptoms are often misdiagnosed as having an allergic reaction or sun allergy. Repeated phototoxic reactions lead to skin thickening and scar formation, with milia being a discrete but distinctive sign of blistering skin diseases (4).
Acquired EPP, unlike its inherited counterpart, tends to occur in adults and is often associated with haematological disorders like myelodysplastic syndrome (MDS) (1, 5–7). The disruption in the ferrochelatase activity due to the underlying MDS leads to protoporphyrin IX accumulation and subsequent photosensitivity. Acquired EPP predominantly affects males, with most cases occurring in adults with a mean age of 58 years. All reported cases were associated with haematological disease, which was affecting chromosome 18q and causing either a deletion or mutation in the FECH gene (1). Clinical presentation of EPP in these cases appears to be more atypical and severe, with cutaneous blister formation and hepatotoxicity, both of which are unusual in heritable EPP (1). Our case confirms more severe cutaneous affliction without hepatic involvement.
Management of EPP, including acquired EPP, focuses on preventing phototoxic reactions and alleviating symptoms. Key strategies include avoiding sunlight, wearing sun-protective clothing, and using broad sunscreens (2). Oral beta-carotene has been used with varying success to provide some photoprotection, particularly in acquired EPP (1). MDS-targeting approaches using azacitidine or bone marrow transplantation should be individually evaluated, depending on the disease severity (1).
A significant advancement in EPP therapy is afamelanotide, a synthetic analogue of alpha-melanocyte-stimulating hormone (α-MSH). Afamelanotide increases melanin production in the skin, providing a natural photoprotective effect by absorbing ultraviolet radiation and visible light. Clinical trials have demonstrated that afamelanotide can significantly reduce the severity and frequency of phototoxic reactions, thereby improving the quality of life for EPP patients (8, 9). Administered as a subcutaneous implant, afamelanotide is generally well tolerated, though it may cause side effects, such as nausea and headache. Two years after the diagnosis of acquired EPP, our patient was able to control his photosensitivity with the above-mentioned sun-protective measures and did not require the use of afamelanotide. To date, he has not developed any signs of liver toxicity despite consistently high levels of plasma porphyrins, which are being monitored on regular basis.
In conclusion, our case underlines the importance of an interdisciplinary approach to evaluate a patient with sudden onset of photosensitivity and an underlying myeloproliferative disorder. While EPP remains a difficult condition to manage, the combination of preventative strategies, innovative therapies like afamelanotide, and vigilant monitoring for complications provides a comprehensive approach to care. This multi-faceted management strategy helps mitigate the impact of the disease, allowing patients to lead more normal and less restricted lives despite the challenges posed by their condition.
REFERENCES
- Snast I, Kaftory R, Sherman S, Edel Y, Hodak E, Levi A, et al. Acquired erythropoietic protoporphyria: a systematic review of the literature. Photodermatol Photoimmunol Photomed 2020; 36: 29–33. https://doi.org/10.1111/phpp.12501
- Erythropoietic Protoporphyria. NCBI Bookshelf. Treasure Island, FL: StatPearls.
- Maitra D, Bragazzi Cunha J, Elenbaas JS, Bonkovsky HL, Shavit JA, Omary MB. Porphyrin-induced protein oxidation and aggregation as a mechanism of porphyria-associated cell injury. Cell Mol Gastroenterol Hepatol 2019; 8: 535–548. https://doi.org/10.1016/j.jcmgh.2019.06.006
- Patsatsi A, Uy CDC, Murrell DF. Multiple milia formation in blistering diseases. Int J Womens Dermatol 2020; 6: 199–202. https://doi.org/10.1016/j.ijwd.2020.03.045
- Yoshioka A, Fujiwara S, Kawano H, Nakano H, Taketani S, Matsui T, et al. Late-onset erythropoietic protoporphyria associated with myelodysplastic syndrome treated with azacitidine. Acta Derm Venereol 2018; 98: 275–277. https://doi.org/10.2340/00015555-2829
- Suzuki H, Kikuchi K, Fukuhara N, Nakano H, Aiba S. Case of late-onset erythropoietic protoporphyria with myelodysplastic syndrome who has homozygous IVS3-48C polymorphism in the ferrochelatase gene. J Dermatol 2017; 44: 651–655. https://doi.org/10.1111/1346-8138.13709
- Blagojevic D, Schenk T, Haas O, Zierhofer B, Konnaris C, Trautinger F. Acquired erythropoietic protoporphyria. Ann Hematol 2010; 89: 743–744. https://doi.org/10.1007/s00277-009-0859-7
- Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey A V, Bissell DM, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med 2015; 373: 48–59. https://doi.org/10.1056/NEJMoa1411481
- Biolcati G, Marchesini E, Sorge F, Barbieri L, Schneider-Yin X, Minder EI. Long-term observational study of afamelanotide in 115 patients with erythropoietic protoporphyria. Br J Dermatol 2015; 172: 1601–1612. https://doi.org/10.1111/bjd.13598