QUIZ SECTION
Persistent Red Skin Nodule on the Dorsum of the Left Hand: A Quiz
Yoshihito MIMA1,2 and Tsutomu OHTSUKA2
1Department of Dermatology, Tokyo Metropolitan Police Hospital, Tokyo, Japan, and 2Department of Dermatology, International University of Health and Welfare Hospital, Tochigi, Japan. E-mail: yoshihito11.mima@gmail.com
Citation: Acta Derm Venereol 2025; 105: adv42133. DOI: https://doi.org/10.2340/actadv.v105.42133.
Copyright: © 2025 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Published: Feb 5, 2025
A 77-year-old man with no history of internal organ disease presented with a 3 cm red nodule on the dorsum of his left hand (Fig. 1). The nodule was asymptomatic and exhibited size fluctuations. Subsequently, he developed systemic symptoms, including fever, malaise, and loss of appetite. Both the cutaneous and systemic symptoms have persisted without regression.
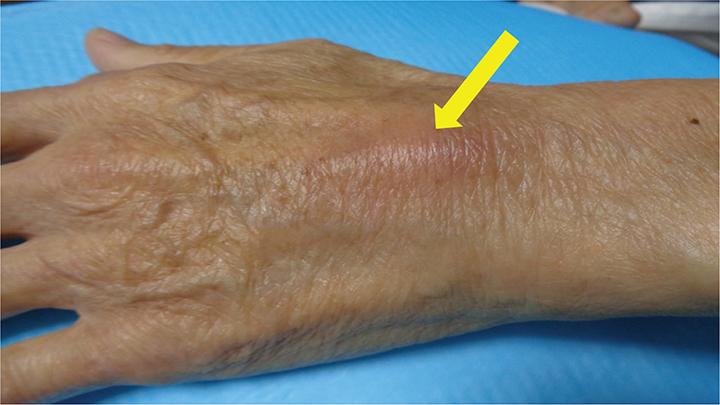
Fig. 1. Clinical photo: a raised red skin nodule measuring 3 cm was observed on the dorsum of the left hand, with mild palpable induration (yellow arrow).
Laboratory tests revealed macrocytic anaemia (Hb 8.9 g/dL, MCV 118 fL), leukopenia (2,100/μL), and thrombocytopenia (9.1×104/μL), indicating pancytopenia. The inflammatory markers C-reactive protein (CRP, 18.2 mg/dL) and lactate dehydrogenase (LDH, 347 U/L) were markedly increased. Tests for tuberculosis, human immunodeficiency virus, hepatitis B, and syphilis were negative. Markers of collagen vascular diseases, including vasculitis, were also negative. Computed tomography revealed pulmonary infiltrates; no other abnormalities were observed. No bacteria were detected in blood cultures.
Histopathological examination of the red single nodule revealed significant perivascular neutrophil infiltration (Fig. 2). No atypical cell infiltrates or epithelioid granulomas were observed. Tissue cultures were negative for fungi and acid-fast bacilli.
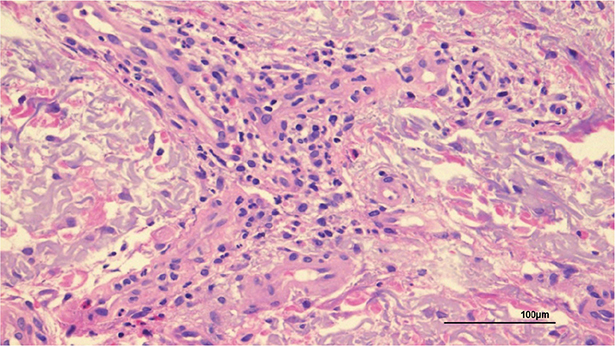
Fig. 2. Histopathological findings of the red nodule: histopathological examination shows significant perivascular neutrophil infiltration (haematoxylin and eosin staining; ×200).
Considering the possibility of haematological malignancies, a bone marrow biopsy was performed, which revealed bone marrow hyperplasia and approximately 7% blasts, suggesting myelodysplastic syndrome (MDS) and vacuolization of the hematopoietic lineage cells, especially erythroid lineage blasts.
What is your diagnosis?
Differential diagnosis 1: VEXAS syndrome
Differential diagnosis 2: Sweet syndrome
Differential diagnosis 3: Erythema nodosum
Differential diagnosis 4: Cutaneous lymphoma
See next page for answer.
ANSWERS TO QUIZ
Persistent Red Nodule on the Dorsum of the Left Hand: A Commentary
Diagnosis: VEXAS syndrome
VEXAS syndrome (vacuoles, E1 enzyme, and X-linked, autoinflammatory, somatic) was proposed by Beck et al. in 2020 (1). VEXAS pathogenesis involves mutations in myeloid-restricted ubiquitin-like modifier-activating enzyme 1 (UBA1), an X-linked gene encoding the ubiquitin-activating enzyme E1. This mutation is somatic, and VEXAS syndrome reportedly manifests in middle-aged to elderly individuals (1). A review of 224 patients with VEXAS syndrome revealed that the majority, 215 (96%), were male (2); as UBA1 is located on the X chromosome, males are more susceptible to the mutation (2). Mutations in UBA1 impair the enzyme’s function, causing myeloid-driven autoinflammation and progressive bone marrow failure. Vacuoles in myeloid and erythroid precursor cells are one of the characteristic features of VEXAS syndrome (1).
VEXAS syndrome is progressive, involves multiple organ systems, and presents mainly with systemic inflammatory and haematological manifestations (1). The main clinical features of VEXAS are skin lesions, non-infectious fever, lung involvement, ocular symptoms, relapsing chondritis, venous thrombosis, lymphadenopathy, and arthralgia (1, 3). Skin manifestations include red papules, red nodules, oedematous erythema, red plaques, palpable purpura, and erythematous purpura with induration (4). Mutations in UBA1 cause autoinflammation by activating autoreactive T cells and increasing B lymphocyte autoantibody production (3). Haematological dysfunction or malignancy is thought to involve UBA1 mutations acting as driver clones for myeloid tumours or other clones in the chronic inflammatory microenvironment, increasing the mutant allele frequency and potentially inducing the development of haematological abnormalities (3). Laboratory findings of VEXAS syndrome reveal pancytopenia, including macrocytic anaemia, elevated inflammatory markers (CRP, LDH), and elevated cytokines such as tumour necrosis factor (TNF), interleukin (IL)-6, and interferon-γ (1,2). Cutaneous histopathological findings of VEXAS syndrome may include leukocytoclastic vasculitis and/or dermal infiltration of neutrophils. Therefore, VEXAS syndrome requires differentiation from various conditions such as Sweet syndrome, relapsing polychondritis, polyarteritis nodosa, and giant cell arteritis, and misdiagnoses are common (4). A review of dermatopathology reports of 64 patients with VEXAS syndrome found that 36% had leukocytoclastic vasculitis, 34% had neutrophilic dermatitis, and 30% had perivascular dermatitis. The p.Met41Leu mutation is associated most frequently with neutrophilic dermal infiltration (82%); the p.Met41Val mutation is associated with vasculitis (55%) (4). Bone marrow biopsies show characteristic cytoplasmic vacuolization of myeloid and erythroid precursors in patients with VEXAS (5).
VEXAS syndrome diagnosis is based on clinical suspicion, supported by bone marrow biopsy findings of cytoplasmic vacuoles in myeloid and erythroid precursor cells, and confirmed by detecting UBA1 gene mutations (5). The p.Met41Thr mutation accounts for most cases; however, other mutations have been implicated in the development of VEXAS syndrome, including p.Met41Leu, p.Met41Val, and Ser56Phe. Ongoing research is investigating the genetic variations associated with this condition (6).
Available treatment options for VEXAS include glucocorticoids, cyclophosphamide, methotrexate, azathioprine, and inhibitors of TNF (infliximab and adalimumab), IL-6 (tocilizumab), CD20 (rituximab), IL-17 (secukinumab), IL-12/IL-23 (ustekinumab), and (ruxolitinib, tofacitinib, and baricitinib) (7). Although over 100 cases of VEXAS syndrome have been reported, randomized controlled trials and retrospective studies have not yet been conducted, and evidence supporting treatment efficacy remains limited (8). Bone marrow precursor cells may carry irreversible somatic mutations in VEXAS syndrome. Therefore, the only treatment designed to permanently correct VEXAS syndrome is haematopoietic stem cell transplantation (HSCT), which reconstructs the haematopoietic and immune systems by removing abnormal bone marrow haematopoietic tissue and transplanting healthy stem cells (7). Despite the treatment being highly effective, treatment-related mortality is a significant risk (7).
In our case, a red nodule appeared on the dorsum of the hand, followed by auricular swelling (Fig. 3) and multiple red nodules on the trunk 2 months later. VEXAS syndrome was considered clinically based on fever with negative blood cultures, elevated inflammatory markers, skin manifestation indicative of leukocytoclastic vasculitis, auricular swelling suggestive of relapsing auricular chondritis, pulmonary infiltrates, pancytopenia, and vacuoles in the hematopoietic lineage cells. However, owing to the patient’s poor general condition caused by VEXAS syndrome and recurrent urinary tract infections, he decided to have the symptoms managed with supportive care without pursuing immunosuppressive treatment or initiating chemotherapy.
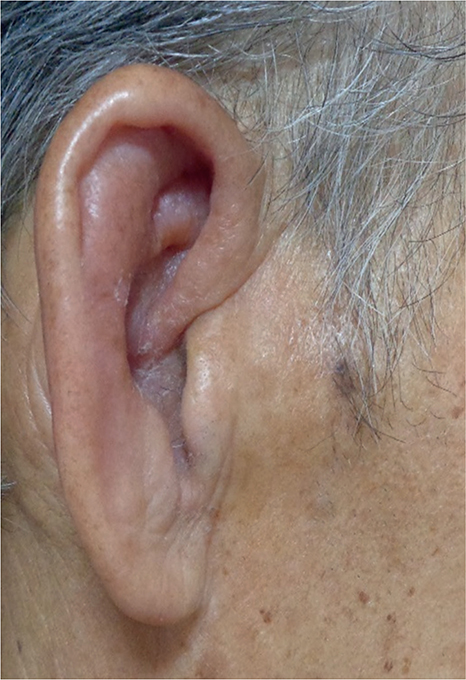
Fig. 3. Clinical photo: two months after the appearance of the rash on the dorsum of the left hand, the marked swelling of bilateral auricles was observed.
UBA1 genetic testing was suggested for the diagnosis of VEXAS syndrome, but the patient declined. Not performing UBA1 genetic testing limited our study. However, a high probability of a UBA1 mutation was suggested based on a scoring system that uses 5 criteria to predict the likelihood of UBA1 mutations (age > 50 years, cutaneous lesions, lung involvement, chondritis, and macrocytic anaemia); our patient met all 5 criteria (5). Patients with the p.Met41Leu variant are thought to have a better prognosis than those with the p.Met41Val variant. This may be attributed to differences in the variations in residual UBA1b translation depending on the type of UBA1 mutation. Clinical manifestations, including skin symptoms, are also suggested to vary based on the type of UBA1 gene mutation (9). Although we were unable to test for the UBA1 mutation in our case, genetic testing should be actively recommended for the early definitive diagnosis of VEXAS syndrome to predict future clinical manifestations and prognosis. Based on these clinical symptoms and prognostic predictions, early consideration of more aggressive interventions, such as HSCT, may become possible (3).
Skin symptoms are often the first sign of VEXAS syndrome, and skin rashes can later emerge with time lags in different regions. VEXAS syndrome has a poor prognosis, possibly because of rapid progression or an inability to initiate immunosuppressive treatments. Therefore, dermatologists can contribute to early diagnosis and treatment by understanding the full clinical course of VEXAS syndrome and carefully observing skin lesions.
REFERENCES
- Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med 2020; 383: 2628–2638.
- Echerbault R, Bourguiba R, Georgin-Lavialle S, Lavigne C, Ravaiau C, Lacombe V. Comparing clinical features between males and females with VEXAS syndrome: data from literature analysis of patient reports. Rheumatology (Oxford) 2024; 63: 2694–2700. https://doi.org/10.1093/rheumatology/keae123
- Loeza-Uribe MP, Hinojosa-Azaola A, Sánchez-Hernández BE, Crispín JC, Apodaca-Chávez E, Ferrada MA, et al. VEXAS syndrome: clinical manifestations, diagnosis, and treatment. Rheumatol Clin (Engl Ed) 2024; 20: 47–56. https://doi.org/10.1016/j.reumae.2023.12.004
- Tan IJ, Ferrada MA, Ahmad S, Fike A, Quinn KA, Groarke EM, et al. Skin manifestations of VEXAS syndrome and associated genotypes. JAMA Dermatol 2024; 160: 822–829. https://doi.org/10.1001/jamadermatol.2024.1657
- Maeda A, Tsuchida N, Uchiyama Y, Horita N, Kobayashi S, Kishimoto M, et al. Efficient detection of somatic UBA1 variants and clinical scoring system predicting patients with variants in VEXAS syndrome. Rheumatology (Oxford) 2024; 63: 2056–2064. https://doi.org/10.1093/rheumatology/kead626
- Al-Hakim A, Savic S. An update on VEXAS syndrome. Expert Rev Clin Immunol 2023; 19: 203–215. https://doi.org/10.1080/1744666X.2023.2157262
- Watanabe R, Kiji M, Hashimoto M. Vasculitis associated with VEXAS syndrome: a literature review. Front Med (Lausanne) 2022; 9: 983939. https://doi.org/10.3389/fmed.2022.983939
- Georgin-Lavialle S, Terrier B, Guedon AF, Heiblig M, Comont T, Lazaro E, et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol 2022; 186: 564–574. https://doi.org/10.1136/annrheumdis-2022-eular.992
- Padureanu V, Marinaș CM, Bobirca A, Padureanu R, Patrascu S, Dascalu AM, et al. Clinical manifestations in Vacuoles, E1 Enzyme, X-Linked, Autoinflammatory, Somatic (VEXAS) syndrome: a narrative review. Cureus 2024; 16: e53041. https://doi.org/10.7759/cureus.53041