SHORT COMMUNICATION
Successful Treatment of Extragenital Lichen Sclerosus with Narrow-band UVB Phototherapy
Lanyu SUN1, Diogo DE SOUSA1  , Pedro DE VASCONCELOS1 , Sónia FERNANDES1 and Paulo FILIPE1–3
, Pedro DE VASCONCELOS1 , Sónia FERNANDES1 and Paulo FILIPE1–3
1Dermatology Department, Hospital de Santa Maria, Unidade Local de Saúde Santa Maria, Av. Prof. Egas Moniz MB, 1649-028 Lisbon, Portugal, 2Faculty of Medicine, Lisbon University, Lisbon, 3Instituto de Medicina Molecular João Lobo Antunes (iMM), Lisbon. E-mail: Lanyusun@gmail.com
Citation: Acta Derm Venereol 2025; 105: adv43470. DOI: https://doi.org/10.2340/actadv.v105.43470.
Copyright: © 2025 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Apr 10, 2025; Accepted after revision: Apr 25, 2025. Published: May 12, 2025.
Competing interests and funding: The authors have no conflicts of interest to declare.
Lichen sclerosus (LS) is a chronic inflammatory skin condition characterized by progressive epidermal atrophy and hypopigmentation, usually affecting the anogenital area. Patients may also present with extragenital lesions, though only 6% of LS cases are isolated to extragenital skin (1, 2). Extragenital lichen sclerosus (EGLS) can be localized or widespread. Given the rare nature of extragenital-only forms of LS, there are limited reports on this entity. Here, we report a patient with widespread EGLS treated successfully with narrowband UV-B (NB-UVB) phototherapy.
CASE REPORT
A 79-year-old woman presented with a 6-month history of a disseminated, severe pruritic eruption. She had no significant medical history. Physical examination revealed widespread shiny plaques, with parchment paper-like atrophy involving mainly the upper trunk, neck, and shoulders (Fig. 1A-C) but also affecting the arms and thighs. No genital involvement was observed. Blood tests including autoimmune panel were normal. Two punch biopsies of the upper back and left forearm showed hyperkeratosis, hypogranulosis, marked epidermal atrophy, and hyalinization of the dermal collagen with sparse perivascular interstitial lymphocytic infiltrate (Fig. 1D). Based on the clinical and histopathological findings, a diagnosis of EGLS was made. She was treated with clobetasol 0.05% cream twice daily and antihistamines, but no improvement was observed after 3 months. The patient underwent 25 sessions, with an irradiation dose ranging from 100 to 2,000 mJ/cm² per session, totalling 29,100 mJ/cm², and an exposure time of 3 to 10 min. Four months after discontinuation of phototherapy, the patient reported complete resolution of pruritus, and a significant improvement in the clinical picture was observed, with the improvement of atrophy and cutaneous surface texture (Fig. 2). No adverse effects were reported during the treatment.

Fig. 1. Clinical and histopathological presentation. (A–C) Well-demarcated, atrophic plaques with a parchment paper-like appearance on the chest, upper back, and left shoulder; (D) marked epidermal atrophy and hyalinization of dermal collagen, with a sparse perivascular interstitial lymphocytic infiltrate (haematoxylin & eosin x100).
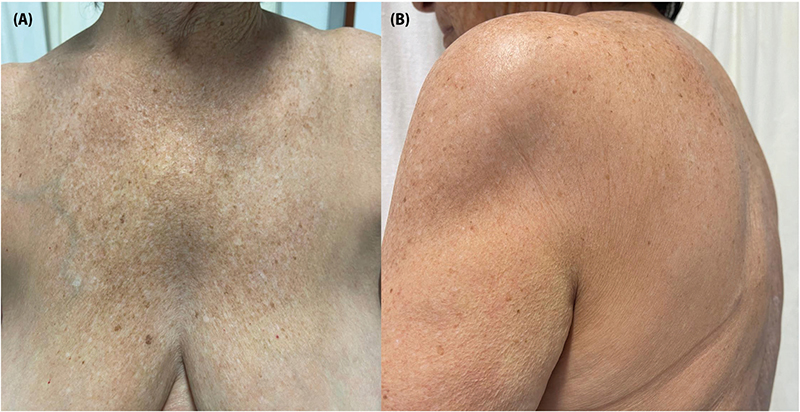
Fig. 2. (A, B) Four months after treatment with narrowband UVB phototherapy. Marked clinical improvement of the lichen sclerosus plaques.
DISCUSSION
LS is a chronic inflammatory disease of unknown aetiology, although genetic, infectious, autoimmune, hormonal, and environmental factors are thought to contribute in its pathogenesis (2, 3). LS has been associated with several autoimmune diseases, including psoriasis, atopic dermatitis, systemic lupus erythematosus, vitiligo, alopecia areata, and Graves’ disease (1). LS is more prevalent in women than in men, with a female-to-male ratio ranging from 6:1 to 10:1. The genital form tends to affect individuals at the extremes of age, with 2 incidence peaks, whereas the extragenital form usually occurs in middle-aged adults (3). A case series of 3 patients with EGLS reported a mean age of 75 years, higher than previously described, similar to our patient (2). In contrast to genital LS, which is often accompanied by severe itching and burning, EGLS is typically asymptomatic (3). However, disease progression may lead to discomfort and pruritus (4), as observed in our patient. Typical lesions of EGLS present as white, opalescent papules, patches, or plaques that slowly progress over time resulting in parchment-like skin – usually affecting the upper trunk, neck, and shoulders (3, 5). The diagnosis of EGLS can be made clinically and confirmed through histopathological examination. Histologic features of EGLS vary depending on disease duration. Early-stage disease is characterized by interface dermatitis with vacuolar/lichenoid changes. As the disease progresses, findings may include follicular plugging, upper dermal sclerosis with a band of hyalinization, and inflammatory infiltrate. Late-stage disease is characterized by epidermal atrophy, loss of rete ridges, upper dermal sclerosis, and a sparse inflammatory infiltrate (3). Ultrapotent topical corticosteroids are considered the first-line treatment. Other topical therapeutic options include topical calcineurin inhibitors, vitamin D analogues, and topical retinoids (3). Extragenital lesions tend to be less responsive to conventional therapy. A recent report described successful treatment of EGLS with ruxolitinib, a new topical Janus kinase (JAK) inhibitor (2). Systemic therapy may be considered for refractory or generalized LS. There are reports of the use of oral corticosteroids, methotrexate, hydroxychloroquine, retinoids, and cyclosporine (3). Phototherapy has been explored as an alternative long-term treatment option for EGLS, with promising results. Topical PUVA, UVA-1, and NB-UVB have been utilized, generally with minimal side effects (3, 6). Kreuter et al. (6) reported 10 cases of EGLS treated with UVA-1 phototherapy, demonstrating good efficacy. We identified 2 case reports describing successful treatment of EGLS with NB-UVB (4, 7). NB-UVB radiation is believed to delay the development of skin sclerosis by decreasing proinflammatory cytokines and increasing the expression of matrix metalloproteinases (5). The efficacy of NB-UVB in sclerosing skin diseases may be limited by its reduced depth of penetration compared with UVA-1. However, this limitation may not apply to LS, which primarily affects the epidermis and superficial dermis (4). The clinical response observed in our patient suggests that NB-UVB therapy may provide therapeutic benefits in EGLS, not only alleviating symptoms but also potentially modifying the disease course. The greater availability of NB-UVB compared with UVA-1, along with its more favourable safety profile, makes it an attractive alternative treatment for EGLS. Further clinical research is needed to better understand the pathogenesis of this rare LS variant and to evaluate the effectiveness of this therapeutic approach.
REFERENCES
- Ezekwe N, Neelam R, Jones BA, Ozog DM, Hamzavi IH. Clinical practice gaps in patients with extragenital lichen sclerosus: a retrospective review. J Am Acad Dermatol 2023; 89: 182–184. https://doi.org/10.1016/j.jaad.2023.02.051
- Jiang C, Muradova E, Lu J. Generalized extragenital lichen sclerosus et atrophicus in skin of color. JAAD Case Rep 2023; 40: 63–66. https://doi.org/10.1016/j.jdcr.2023.07.031
- Arif T, Fatima R, Sami M. Extragenital lichen sclerosus: a comprehensive review. Australas J Dermato 2022; 63: 452–462. https://doi.org/10.1111/ajd.13890
- Colbert RL, Chiang MP, Carlin CS, Fleming M. Progressive extragenital lichen sclerosus successfully treated with narrowband UV-B phototherapy. Arch Dermatol 2007; 143: 19–20. https://doi.org/10.1001/archderm.143.1.19
- Burshtein A, Burshtein J, Rekhtman S. Extragenital lichen sclerosus: a comprehensive review of clinical features and treatment. Arch Dermatol Res 2023; 315: 339–346. https://doi.org/10.1007/s00403-022-02397-1
- Kreuter A, Gambichler T, Avermaete A, Happe M, Bacharach-Buhles M, Hoffmann K, et al. Low-dose ultraviolet A1 phototherapy for extragenital lichen sclerosus: results of a preliminary study. J Am Acad Dermatol 2002; 46: 251–255. https://doi.org/10.1067/mjd.2002.118552
- de Brito FF, de Andrade TCPC, Coelho APCP, Pinto ACVD, Nunes AJF, Tonello CS. Successful treatment of a rare case of widespread extragenital lichen sclerosus with narrow band UVB phototherapy. Surg Cosmet Dermatol 2015; 7: 59–62. https://doi.org/10.5935/scd1984-8773.2015731668