ORIGINAL REPORT
Rapidly Progressing CD8-negative Hypopigmented Mycosis Fungoides in Adult Caucasian Male with Good Response to Mogamulizumab
Agnieszka KIMAK-PIELAS1,2, Tadeusz ROBAK3,4, Marcin BRAUN5, Agnieszka ŻEBROWSKA1,2 and Ewa ROBAK1,2
1Department of Dermatology, Medical University of Lodz, Łódź, 2Department of Dermatology and Venereology, Teaching Hospital No 2, Łódź, 3Department of Haematology, Medical University of Łódź, Lodz, 4Department of General Haematology, Copernicus Memorial Hospital, Łódź, and 5Department of Pathology, Medical University of Lodz, Łódź, Poland
Hypopigmented mycosis fungoides is a rare variant of mycosis fungoides often seen in younger patients and individuals with darker skin tones and is characterized by hypopigmented patches or plaques. The lesions are usually asymptomatic and respond well to topical treatment or phototherapy. This article presents a case of an adult Caucasian male with a rare variant of CD8-negativeh hypopigmented mycosis fungoides, with a rapid progression to erythrodermic lesions, failure of standard treatment, and a good response to mogamulizumab.
SIGNIFICANCE
Hypopigmented mycosis fungoides is a rare form of skin cancer that usually appears as light patches on the skin, most commonly in younger people or those with darker skin tones, and typically can be treated with ointments or light therapy. However, in some cases the condition can worsen rapidly, leading to more severe symptoms and resistance to standard treatments. In this study, we present a case of a Caucasian man who developed this rare form of skin cancer later in life, which progressed quickly and did not respond to the usual treatments.
Key words: hypopigmented mycosis fungoides; Caucasian; mogamulizumab
Citation: Acta Derm Venereol 2025; 105: adv43932. DOI: https://doi.org/10.2340/actadv.v105.43932.
Copyright: 2025 © The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: May 23, 2025. Accepted after revision: Nov 24, 2025. Published: Dec 18, 2025.
Corr: Agnieszka Kimak-Pielas, Department of Dermatology, Medical University of Lodz, Łódź, Poland. E-mail: Agnieszka.kimak@umed.lodz.pl
Competing interests and funding: The authors have no conflicts of interest to declare.
This research was funded by the Medical University of Lodz under grant number 503/1-152-01/503-11-002.
INTRODUCTION
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma (CTCL). The hypopigmented variant is rare and often misdiagnosed. It occurs more frequently in children, adolescents, and young adults. Higher prevalence of hypopigmented mycosis fungoides (HMF) is observed in individuals with darker phototypes – Fitzpatrick skin types IV–VI (1). Lesions usually develop on the trunk and proximal parts of the extremities and present as vitiligo-like macules, patches, or plaques, sometimes with fine scaling and mild pruritus. In some cases, erythematous lesions may coexist with hypopigmented ones. Lesions might resemble parapsoriasis, atopic dermatitis, pityriasis alba, pityriasis lichenoides chronica, vitiligo, tinea versicolor, leprosy (particularly in endemic areas), sarcoidosis, and post-inflammatory hypopigmentation, which can pose a diagnostic challenge and lead to misdiagnosis (1–3).
Diagnosis is based on histopathology examination with common present features including lymphocytic infiltration in the epidermis (Pautier’s microabscesses) and upper dermis with variable epidermal changes including focal parakeratosis, acanthosis, and spongiosis, as well as melanin loss in basal keratinocytes with reduced melanocytes. Immunohistochemistry and T-cell receptor gene rearrangement test may be utilized to confirm clonal proliferation. Assessment of lymph nodes, blood, and systemic involvement is required for staging the disease (1, 4, 5).
In classic MF a predominance of CD4+ T-cells is typically seen. In contrast, the pathogenesis of HMF involves clonal expansion of malignant T-cells with a predominance of CD8+ cytotoxic T-cells. The role of CD8+ T-cells remains unclear and opposing roles are suggested, both mediated by their cytotoxic mechanisms. On the one hand, malignant CD8+ cells infiltrate dermis and epidermis, and release cytotoxic molecules that cause inflammation, damage melanocytes, and disrupt melanin production. As a result, hypopigmented skin lesions develop. On the other hand, CD8+ T-cells may also have a tumour-suppressing effect, by recognizing and eliminating malignant clones of T-cells. This may explain better prognosis in HMF compared with the classic variant. The balance between these roles remains unclear and perhaps depends on other immune cells and cytokine signalling, forming the tumour microenvironment (1, 6).
The HMF course is typically indolent, skin lesions rarely exceed stage IB, and treatment is usually focused on skin-directed therapies. Skin lesions respond well to light exposure and narrowband ultraviolet B (NB-UVB) as well as psoralen plus ultraviolet A (PUVA) phototherapy. In first-line topical treatment corticosteroids or nitrogen mustard are implemented. Refractory or advanced cases are rare and sometimes retinoids, interferons, or methotrexate should be considered. Although HMF has an indolent course and a good prognosis, progression to more advanced stages with involvement of lymph nodes, blood, or internal organs can occur in some cases. Thus, regular monitoring for disease progression or transformation to a more aggressive form of CTCL is crucial for optimizing outcomes of patients with HMF (1).
We describe the case of an adult Caucasian man who initially presented with CD8-negative hypopigmented mycosis fungoides, which later evolved into an erythrodermic variant. The disease proved refractory to standard therapies but demonstrated a favourable response to mogamulizumab. The presented case underscores the importance of close monitoring in MF patients, irrespective of their clinical presentation, and suggests a new possible treatment options for advanced cases.
CASE REPORT
A 48-year-old generally healthy Fitzpatrick II male, over 6 months prior to admission developed hypopigmented patches with fine scaling and mild pruritus predominantly located on the groins and armpits, with concomitant erythematous macules (Fig. 1A). Skin biopsies from both vitiligo-like and erythematous macules were performed and both showed subepidermal infiltration by medium-sized and small lymphoid cells with subtle atypia, demonstrating epidermotropism and the formation of Pautrier microabscesses, and the same immunophenotype: CD3+, CD4+, CD8–, CD7– (Fig. 2). The patient was diagnosed with hypopigmented mycosis fungoides, and because the disease is normally slow-progressing, a short course of systemic alongside topical steroids with UVA-1 phototherapy was initiated.
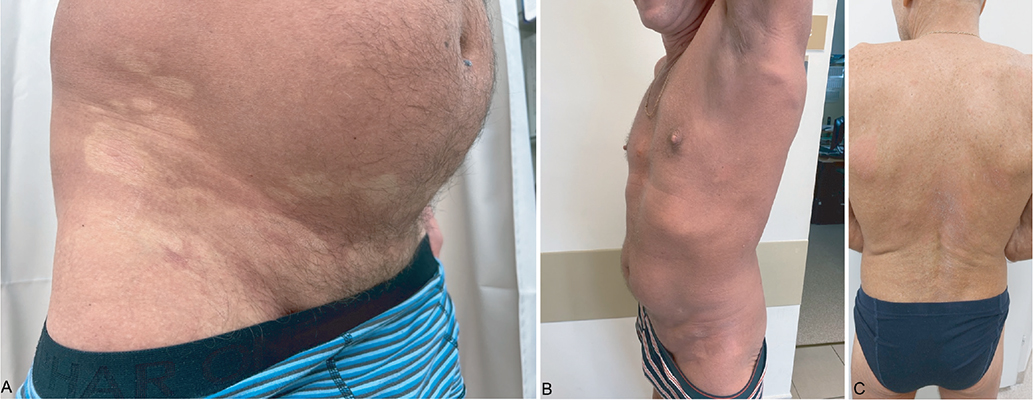
Fig. 1. Clinical pictures of the patient. (A) Hypopigmented and erythematous skin lesions with fine scaling in the initial phase. (B) Erythroderma with lymphadenopathy at a later stage of the disease. (C) New infiltrative plaques on the back; loss of response to mogamulizumab.
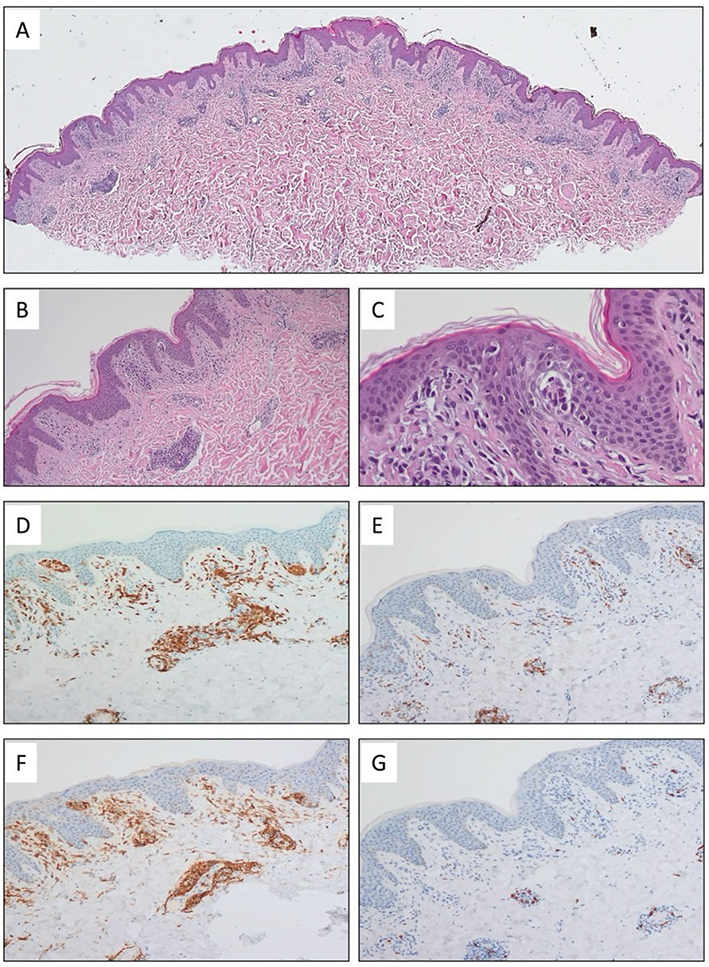
Fig. 2. Histopathological images of skin affected by mycosis fungoides. (A–C) Haematoxylin and eosin (H&E) staining reveals a skin biopsy with subepidermal infiltration by medium-sized and small lymphoid cells with subtle atypia, demonstrating epidermotropism and the formation of Pautrier microabscesses. Magnifications: 25x (A), 100x (B), and 400x (C). (D–G) In immunohistochemistry, the lymphoid cells were CD3-positive (D, 100x). CD7 expression was notably decreased (E, 100x), and staining for CD4 and CD8 showed a significant predominance of CD4-positive cells (F–G, 100x).
After 3 months of skin-directed therapy, the patient developed confluent infiltrative erythematous lesions with a slightly orange hue. The lesions were covering over 80% of the body surface area and were accompanied by intense itch, hyperkeratosis of the palms and soles, and peripheral lymphadenopathy of axillary, inguinal, head and neck lymph nodes (Fig. 1B).
A biopsy from an enlarged inguinal lymph node showed effacement of the lymph node architecture by diffuse infiltration of atypical medium-sized lymphoid cells, with immunophenotype consistent with the skin samples (Fig. 3). Peripheral blood flow cytometry showed <5% atypical cells, a CD4/CD8 ratio of 3,8, a CD4+CD7- ratio of 0.9%, and a CD4+CD26- ratio of 2.3%. Bone marrow biopsy was normocellular. In the PET/CT scan of the head and neck, chest, abdomen, and pelvic region, metabolically active and enhanced foci were visualized in the skin and subcutaneous tissue, bilateral axillary, inguinal, and external iliac lymph nodes, and 1 right common iliac lymph node. There was no involvement of visceral organs. TCR gene rearrangement studies could not be conducted because of resource constraints. Eventually the patient was diagnosed with erythrodermic MF originating from hypopigmented mycosis fungoides T4N3M0B0, clinical stage IVA2.
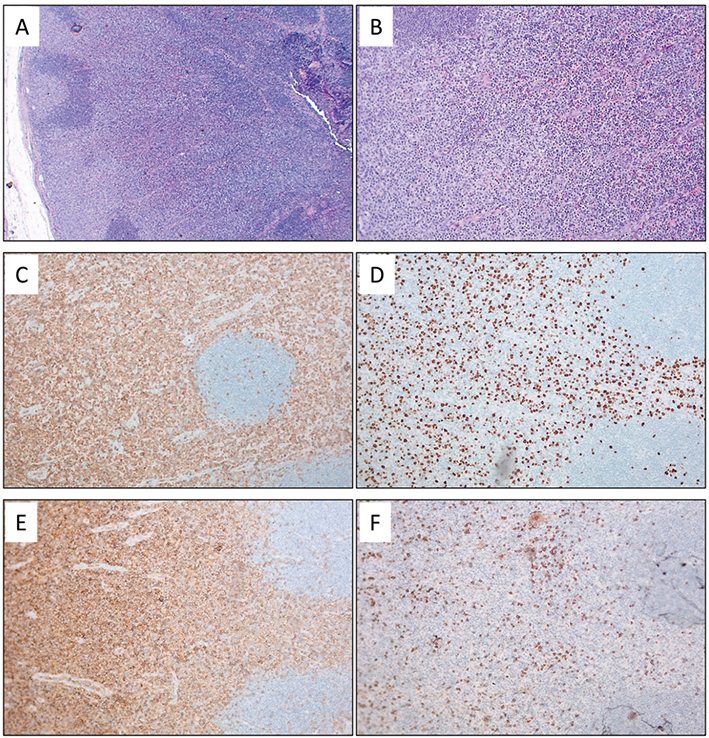
Fig. 3. Histopathological images of a lymph node affected by mycosis fungoides. (A-B): Haematoxylin and eosin (H&E) staining shows effacement of the lymph node architecture by diffuse infiltration of atypical medium-sized lymphoid cells, with preservation of lymphoid follicles. Magnifications: 25x (A) and 100x (B). (C–F) In immunohistochemistry, the lymphoid cells were CD3-positive (C, 100x), with an increased Ki67 proliferation index (D, 100x). Staining for CD4 and CD8 revealed a significant predominance of CD4- positive cells (E–F, 100x). The histopathological findings were consistent between the skin (Fig. 1) and the lymph node.
Treatment with methotrexate at a dose of 25 mg per week, alongside clobetasol, was initiated. Although partial remission of skin lesions and pruritus was initially achieved, the disease progressed even after increasing the methotrexate dose to 30 mg per week. Treatment with methotrexate was ceased after 7 months. The patient was then given mogamulizumab (MOGA) 1 mg/kg i.v., administered in 28-day cycles: in the first cycle on days 1, 8, 15, and 22, and in subsequent cycles on days 1 and 15. The patient achieved almost complete response to MOGA, which was lost after 6 months, when new infiltrative plaques gradually appeared on the trunk (Fig. 1C). Then, methotrexate at a dose of 20 mg per week p.o. was added to MOGA treatment and the combined therapy had now been ongoing for 5 months resulting in partial remission. Hypopigmented, infiltrative, and a majority of erythematous lesions, as well as peripheral lymphadenopathy, resolved. Occasionally, new erythematous macules occur. The patient has not experienced any side effects apart from asymptomatic moderate neutropenia. The treatment is planned to continue until side effects occur or response is lost.
DISCUSSION
Hypopigmented mycosis fungoides (HMF) is most frequently observed in younger individuals, although the age of symptom onset ranges from early childhood to over 70 years (1, 4, 7). HMF is typically indolent; however, in some instances it may progress to tumour-stage disease or erythroderma. Despite potential responsiveness to treatment, certain cases can ultimately prove fatal (4, 8).
The role of CD8+ T-cells in the pathogenesis of HMF remains unclear. It is claimed that these neoplastic cells exert a cytotoxic effect towards melanocytes and decrease their expression of CD117 and secretion of the microphthalmia-associated transcription factor (MITF) (5, 9–11). These 2 molecules interact with each other and control melanin production. Similarly to vitiligo, change in their expression results in hypopigmented skin lesions formation, and lesions repigmentation suggests treatment effectiveness.
While predominant epidermotropism of CD8+/ lymphocytes is considered a hallmark of hypopigmented mycosis fungoides (1, 12), cases exhibiting a CD4+/ CD8– immunophenotype have also been documented (4, 13–15). The course of CD4+ HMF is heterogeneous, ranging from stage IA disease responsive to topical or UV therapy (14, 15), to aggressive progression complicated by bone marrow aplasia and fatal septicaemia (13). Moreover, CD4/CD8 double-negative immunophenotype HMF has also been reported in the literature (16, 17). The reported cases did not exceed stage II, and responded well to a combination of narrow band ultraviolet B and topical corticosteroid therapy. According to available reviews, CD4/CD8 double-negative mycosis fungoides tends to occur in younger individuals and is often associated with atypical clinical presentations, particularly hypopigmented variants (16).
Mogamulizumab (MOGA) is a humanized monoclonal antibody targeting C-C chemokine receptor 4 (CCR4) expressed on T-cells and particularly overexpressed on malignant T-cells, such as in mycosis fungoides (MF) and Sézary syndrome (SS). CCR4 is also expressed on the surface of T-regulatory cells, which may promote an immunosuppressive tumour microenvironment. Mogamulizumab’s mechanism of action combines antibody-directed cellular cytotoxicity and immune modulation by depletion of regulatory T cells (18, 19).
The primary evidence supporting mogamulizumab’s use comes from the MAVORIC trial (a phase III, open-label, randomized study), which demonstrated its efficacy in patients with MF and SS (20). MOGA was established as a superior treatment option compared with vorinostat. However, higher response rates were observed for SS than MF (37% vs 21%). Notably, enrolment to this trial did not require CCR4 expression (21). However, CCR4 is more commonly expressed in Sézary syndrome, which may partially explain the higher response rates observed in SS patients. In cases with lower expression of CCR4, impact on T regs or the tumour microenvironment might be attributed to the therapeutic effect (19).
In contrast to classic mycosis fungoides, the hypopigmented variant is typically characterized by CD8+ T cells, which often lack CCR4 expression (2, 22). This raises the question of the potential benefit of mogamulizumab in HMF. Giving the limited data addressing its effectiveness in CD8+ HMF, its role remains uncertain. Our patient presented with a rare variant of CD8- HMF and exhibited a remarkable response to mogamulizumab. Further studies are warranted to better define the role of mogamulizumab in patients with atypical immunophenotypes of mycosis fungoides.
The presented case is unusual for several reasons. First, hypopigmented mycosis fungoides (HMF) developed in an adult Caucasian patient, whereas it typically affects children and young individuals with darker skin phototypes. Second, the immunophenotype was CD8- negative, which is atypical, as HMF usually exhibits CD8+ predominance. Third, the disease followed an unusually aggressive course and we suggest that it may be related to the depletion of CD8+ tumour-infiltrating cells. Additionally, treatment with mogamulizumab was initiated, resulting in a favourable response. To our knowledge, this is the first reported case of HMF successfully treated with mogamulizumab. The presented case underscores the importance of close monitoring in mycosis fungoides patients, irrespective of their clinical presentation, and suggests a new possible treatment option for advanced cases.
ACKNOWLEDGEMENTS
Informed consent statement: Written informed consent has been obtained from the patient to publish this paper.
REFERENCES
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol 2013; 88: 954. https://doi.org/10.1590/ABD1806-4841.20132336
- Qari MS, Li N, Demierre M-F. Hypopigmented mycosis fungoides: case reports and literature review. J Cutan Med Surg 2000; 4: 142–148. https://doi.org/10.1177/120347540000400306
- Djawad K. Hypopigmented mycosis fungoides mimicking leprosy successfully treated with oral and topical corticosteroids: a new great imitator? Acta Dermatovenerol Alp Pannonica Adriat 2021; 30: 83–85. https://doi.org/10.15570/actaapa.2021.20
- Ardigó M, Borroni G, Muscardin L, Kerl H, Cerroni L. Hypopigmented mycosis fungoides in Caucasian patients: a clinicopathologic study of 7 cases. J Am Acad Dermatol 2003; 49: 264–270. https://doi.org/10.1067/S0190-9622(03)00907-1
- Rodney IJ, Kindred C, Angra K, Qutub ON, Villanueva AR, Halder RM. Hypopigmented mycosis fungoides: a retrospective clinicohistopathologic study. J Eur Acad Dermatol Venereol 2017; 31: 808–814. https://doi.org/10.1111/JDV.13843
- Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest 2005; 115: 798–812. https://doi.org/10.1172/JCI24826
- Shi HZ, Jiang YQ, Xu XL, Zhang W, Song H, Wang XP, et al. Hypopigmented mycosis fungoides: a clinicopathological review of 32 patients. Clin Cosmet Investig Dermatol 2022; 15: 1259–1264. https://doi.org/10.2147/CCID.S370741
- Stone ML, Styles AR, Cockerell CJ, Pandya AG. Hypopigmented mycosis fungoides: a report of 7 cases and review of the literature. Cutis 2001; 67: 133–138.
- Singh ZN, Tretiakova MS, Shea CR, Petronic-Rosic VM. Decreased CD117 expression in hypopigmented mycosis fungoides correlates with hypomelanosis: lessons learned from vitiligo. Mod Pathol 2006; 19: 1255–1260. https://doi.org/10.1038/MODPATHOL.3800644
- Kitamura R, Tsukamoto K, Harada K, Shimizu A, Shimada S, Kobayashi T, et al. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis: role of SCF/KIT protein interactions and the downstream effector, MITF-M. J Pathol 2004; 202: 463–475. https://doi.org/10.1002/PATH.1538
- El-Darouti MA, Marzouk SA, Azzam O, Fawzi MM, Abdel-Halim MR, Zayed AA, et al. Vitiligo vs. hypopigmented mycosis fungoides (histopathological and immunohistochemical study, univariate analysis). Eur J Dermatol 2006; 16: 17–22.
- El Shabrawi-Caelen L, Cerroni L, Medeiros LJ, McCalmont TH. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol 2002; 26: 450–457. https://doi.org/10.1097/00000478-200204000-00006
- Sigal M, Grossin M, Laroche L, Basset F, Aitken G, Haziza JL, et al. Hypopigmented mycosis fungoides. Clin Exp Dermatol 1987; 12: 453–454. https://doi.org/10.1111/j.1365-2230.1987.tb01948.x
- Robert C, Moulonguet I, Baudot N, Flageul B, Dubertret L. Hypopigmented mycosis fungoides in a light-skinned woman [2]. Br J Dermatol 1998; 139: 341–343. https://doi.org/10.1046/j.1365-2133.1998.02379.x
- Amichai B, Grunwald MH, Avinoach I, Halevy S. Hypopigmented mycosis fungoides in a white female. J Dermatol 1996; 23: 425–426. https://doi.org/10.1111/j.1346-8138.1996.tb04046.x
- Ito A, Sugita K, Ikeda A, Yamamoto O. CD4/CD8 double-negative mycosis fungoides: a case report and literature review. Yonago Acta Med 2019; 62: 153–158. https://doi.org/10.33160/yam.2019.03.021
- Kempf W, Kazakov DV, Cipolat C, Kutzner H, Roncador G, Tomasini D. CD4/CD8 double negative mycosis fungoides with PD-1 (CD279) expression: a disease of follicular helper T-cells? Am J Dermatopathol 2012; 34: 757–761. https://doi.org/10.1097/DAD.0B013E31825B26D1
- Poteligeo. Product information. Available on Dec 8, 2025 https://www.ema.europa.eu/en/documents/product-information/poteligeo-epar-product-information_en.pdf
- Fernández-Guarino M, Ortiz P, Gallardo F, Llamas-Velasco M. Clinical and real-world effectiveness of mogamulizumab: a narrative review. Int J Mol Sci 2024; 25: 2203. https://doi.org/10.3390/IJMS25042203
- Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol 2018; 19: 1192–1204. https://doi.org/10.1016/S1470-2045(18)30379-6
- Rao M, Young K, Jackson-Cowan L, Kourosh A, Theodosakis N. Post-inflammatory hypopigmentation: review of the etiology, clinical manifestations, and treatment options. J Clin Med 2023; 12: 1243. https://doi.org/10.3390/JCM12031243
- Rao AG, Naresh M, Shreeja V, Vsvn S, Sindhuja B, Pranaya B. CD4/CD8 dual positive pityriasis lichenoides like mycosis fungoides presenting with both pityriasis lichenoid lesions and hypopigmented patches: a rare presentation. Indian J Dermatol 2022; 67: 441–444. https://doi.org/10.4103/ijd.ijd_279_22