RESEARCH LETTER
Characterization of the Gut Microbiota in Patients with Erythroderma
Huizhong WANG1-3#, Ruojun WANG1-3#, Yang WANG1-4* and Jingru SUN1-3*
1Department of Dermatology and Venereology, Peking University First Hospital, Beijing, 2National Clinical Research Center for Skin and Immune Diseases, Beijing, 3NMPA Key Laboratory for Quality Control and Evaluation of Cosmetics, Beijing, and 4Peking-Tsinghua Center for Life Sciences, Peking University, Beijing, China. *E-mails: yangwang_dr@bjmu.edu.cn; sjr12315@126.com
#These authors contributed equally.
Citation: Acta Derm Venereol 2025; 105: adv44643. DOI: https://doi.org/10.2340/actadv.v105.44643.
Copyright: © 2025 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Aug 17, 2025. Accepted after revision: Nov 24, 2025. Published: Dec 16, 2025.
Competing interests and funding: The authors have no conflicts of interest to declare.
This study was supported by the Capital’s Funds for Health Improvement and Research (No. 2024-1-4074) and the National Natural Science Foundation of China (82002903, 82425050).
To the Editor,
Erythroderma is a severe syndrome characterized by widespread erythema and scaling, which may arise from various aetiologies (1). Erythrodermic cutaneous T-cell lymphoma (ECTCL) represents the most aggressive manifestation within this spectrum, frequently associated with systemic involvement and a poor prognosis (1). Conversely, another extremity of this spectrum may stem from prior inflammatory dermatoses, such as atopic dermatitis and psoriasis (1). Previous studies have paid limited attention to the gut microbiome in the context of erythroderma. Therefore, our study aimed to characterize the gut microbiota profiles of patients with erythroderma, particularly focusing on the specific signatures of the gut microbiome associated with ECTCL.
We conducted a cross-sectional analysis of the gut microbiota from 16 erythrodermic atopic dermatitis (EAD) patients, 12 ECTCL patients, and 14 healthy controls. The diagnoses of EAD and ECTCL were based on integrated clinical-pathological criteria and laboratory findings. The exclusion criteria included use of systemic immunomodulators or antibiotics, extreme diets (e.g., strict ketogenic, pure vegan, and extreme macronutrient diets), pregnancy, and intake of prebiotics or probiotics in the previous 4 weeks. Microbial 16S ribosomal RNA genes from the collected stool samples were analysed using third-generation PacBio sequencing technology (2). Differences in intragroup diversity (β-diversity) and taxonomic composition among the groups were assessed using Kruskal–Wallis and Wilcoxon tests. Additionally, the gut microbiota signatures of ECTCL and EAD were established by linear discriminant analysis effect size (LEfSe) and random forest model analysis.
The mean ages of the ECTCL group, EAD group, and control group were 50.6 ± 17.4 years, 59.2 ± 15.5 years, and 55.9 ± 11.8 years, respectively (Table I). No significant differences were found in terms of age, sex, or body mass index among the groups. Analysis of the β-diversity index showed that the ECTCL group exhibited significantly lower intragroup diversity compared with both the control and EAD groups (p < 0.001). Moreover, the non-metric multidimensional scaling (NMDS) plot illustrated a reduced dispersion of the ECTCL group relative to the other 2 groups (Fig. 1A). Bacteroides emerged as the most prevalent genus among all participants (Fig. 1B). The abundance of Faecalibacterium tended to be lower in all erythroderma patients (Control: 14.0%, EAD: 8.6%, ECTCL: 9.2%; EAD vs Control: p > 0.05, ECTCL vs Control: p > 0.05). Importantly, the relative abundance of the Christensenellaceae R.7 group was significantly higher in ECTCL patients than in both control and EAD patients (p < 0.05).
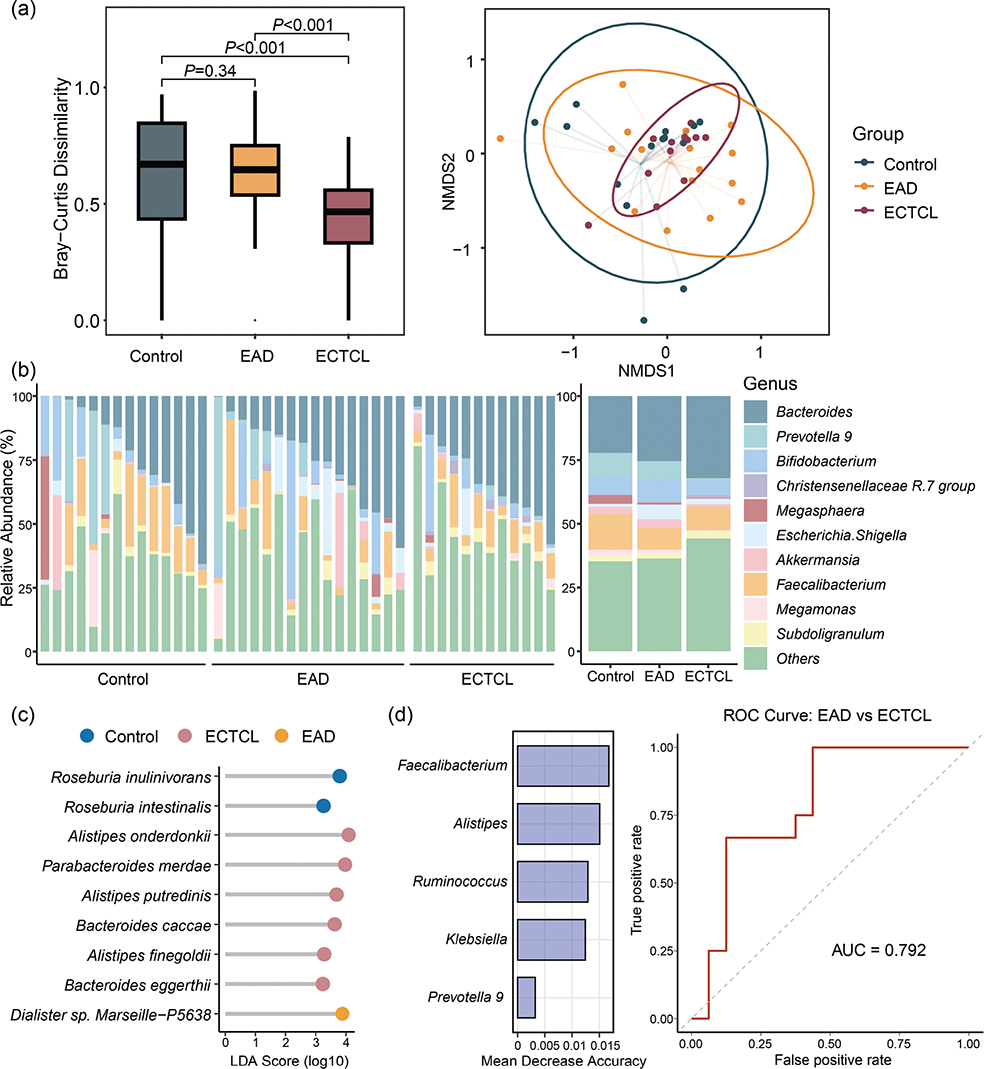
Fig. 1. Features of the gut microbiota in patients with erythroderma and healthy controls. (A) Multidimensional scaling (MDS) plot of gut microbial communities based on Bray–Curtis dissimilarity analysis at the genus taxonomic level revealed significant differences between erythrodermic cutaneous T-cell lymphoma (ECTCL) patients and healthy controls (or erythrodermic atopic dermatitis [EAD] patients). (B) Relative abundance (%) of the 10 most abundant genera in gut microbial communities among healthy controls, EAD patients, and ECTCL patients. (C) LEfSe analysis identified differentially abundant bacteria species among the healthy controls, EAD patients, and ECTCL patients. (D) The mean decreased accuracy (MDA) of the top 5 bacterial genera and receiver operating characteristic (ROC) curves were plotted to discriminate ECTCL from EAD.
LEfSe analysis (Fig. 1C) indicated that Roseburia inulinivorans and Roseburia intestinalis were the predominant bacterial species in healthy controls. Additionally, the species Alistipes onderdonkii, Parabacteroides merdae, Alistipes putredinis, Bacteroides caccae, Alistipes finegoldii, and Bacteroides eggerthii were significantly enriched in ECTCL, most of which are associated with the development of gastrointestinal cancer (3). To further identify ECTCL-specific microbial biomarkers, a random forest model analysis was conducted (Fig. 1D). In total, 5 bacterial genera – Faecalibacterium, Alistipes, Ruminococcus, Klebsiella, and Prevotella 9 – were selected for differentiating ECTCL from EAD, yielding an area under the curve of 0.792.
Our results indicated a non-significant decreasing trend in beneficial gut microorganisms, including Faecalibacterium, in both EAD and ECTCL. Although this trend did not reach statistical significance, lower abundances of Faecalibacterium have previously been associated with dysregulated cytokine signatures characteristic of AD and colorectal cancer (CRC) (4). Moreover, Roseburia intestinalis, which was not prevalent among patients with EAD and ECTCL, has been shown to regulate barrier homeostasis, immune cell function, and cytokine release, primarily through its metabolite butyrate (5). Given that ECTCL and EAD are characterized by T helper 2-predominant cytokine profiles, the dysbiotic signatures may be attributed to the complex interactions between the gut microbiome and the immune milieu (6).
ECTCL exhibited distinct malignant signatures in the gut microbiota compared with EAD. Certain bacterial subpopulations are known to be associated with oncogenesis or influence oncogenesis through immune modulation. The Christensenellaceae R.7 group was predominant in ECTCL and has been reported to be abundant in the gut microbiota of lung cancer patients resistant to therapy (7). Furthermore, Alistipes has been implicated as a potential pathogen in the pathogenesis of CRC (3). Moschen et al. demonstrated that Alistipes finegoldii promotes right-sided colorectal cancer via the IL-6/STAT3 pathway (8). However, further research into the influence of these bacterial subpopulations on the progression of CTCL is warranted.
To our knowledge, this study is the first to characterize the gut microbiome in patients with erythroderma, particularly highlighting the unique malignant signature of gut dysbiosis associated with ECTCL. Given that both EAD and ECTCL exhibit similar clinical manifestations and immune profiles, our findings offer a non-invasive approach for helping differentiate ECTCL from EAD through machine learning techniques based on gut microbiome analysis. However, due to the limited sample size, validation in larger, multicentre cohorts is essential for future investigations.
ACKNOWLEDGEMENTS
IRB approval status: This study was approved by the Biomedical Research Ethics Committee; approval #No. 2023KY297.
REFERENCES
- Pang Y, Nguyen WQ, Guerrero LI, Chrisman LP, Hooper MJ, McCarthy MC, et al. Deciphering the etiologies of adult erythroderma: an updated guide to presentations, diagnostic tools, pathophysiologies, and treatments. Am J Clin Dermatol 2024; 25: 927–950. https://doi.org/10.1007/s40257-024-00886-9
- Buetas E, Jordán-López M, López-Roldán A, D’Auria G, Martínez-Priego L, De Marco G, et al. Full-length 16S rRNA gene sequencing by PacBio improves taxonomic resolution in human microbiome samples. BMC Genomics 2024; 25: 310. https://doi.org/10.1186/s12864-024-10213-5
- Parker BJ, Wearsch PA, Veloo ACM, Rodriguez-Palacios A. The genus Alistipes: gut bacteria with emerging implications to inflammation, cancer, and mental health. Front Immunol 2020; 11: 906. https://doi.org/10.3389/fimmu.2020.00906
- Martin R, Rios-Covian D, Huillet E, Auger S, Khazaal S, Bermudez-Humaran LG, et al. Faecalibacterium: a bacterial genus with promising human health applications. FEMS Microbiol Rev 2023; 47: fuad039. https://doi.org/10.1093/femsre/fuad039
- Kang X, Liu C, Ding Y, Ni Y, Ji F, Lau HCH, et al. Roseburia intestinalis generated butyrate boosts anti-PD-1 efficacy in colorectal cancer by activating cytotoxic CD8(+) T cells. Gut 2023; 72: 2112–2122. https://doi.org/10.1136/gutjnl-2023-330291
- Hooper MJ, LeWitt TM, Pang Y, Veon FL, Chlipala GE, Feferman L, et al. Gut dysbiosis in cutaneous T-cell lymphoma is characterized by shifts in relative abundances of specific bacterial taxa and decreased diversity in more advanced disease. J Eur Acad Dermatol Venereol 2022; 36: 1552–1563. https://doi.org/10.1111/jdv.18125
- Chen H-H, Wu Q-J, Zhang T-N, Zhao Y-H. Gut microbiome and serum short-chain fatty acids are associated with responses to chemo- or targeted therapies in Chinese patients with lung cancer. Front Microbiol 2023; 14: 1165360. https://doi.org/10.3389/fmicb.2023.1165360
- Moschen AR, Gerner RR, Wang J, Klepsch V, Adolph TE, Reider SJ, et al. Lipocalin 2 protects from inflammation and tumorigenesis associated with gut microbiota alterations. Cell Host Microbe 2016; 19: 455–469. https://doi.org/10.1016/j.chom.2016.03.007