SHORT COMMUNICATION
Vedolizumab-induced Bullous Pemphigoid: A Case Report with Serological Evidence and Mechanistic Insights
Xun FENG1†, Jishu LI1†, Xiaohong LI2, Yan ZHANG3 and Wei LI1*
1Department of Dermatology & Venerology, Rare Diseases Center, West China Hospital, Sichuan University, Chengdu, China, 2Department of Outpatient, West China Hospital, Sichuan University, Chengdu, China, and 3Department of Gastroenterology, West China Hospital of Sichuan University, Chengdu, China. *Email: liweihx_hxyy@scu.edu.cn
Citation: Acta Derm Venereol 2026; 106: adv-2025-0090. DOI: https://doi.org/10.2340/actadv.v106.adv-2025-0090.
Copyright: © 2026 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Oct 9, 2025. Accepted after revision: Feb 4, 2026.
Published: Mar 2, 2026.
Competing interests and funding: The authors have no conflicts of interest to declare.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Pemphigoid diseases are a group of subepidermal autoimmune blistering disorders. Patients with pemphigoid often present with blisters on an erythematous or urticarial background, subepidermal bullae with eosinophils or neutrophils on histopathological examination and linear deposition of IgG and/or C3 at the dermo-epidermal junction. Serum tests may also identify specific autoantibodies to basement membrane zone antigens, such as BP180 (1). Numerous cases of drug-induced pemphigoid, including reactions to certain biological agents, have been reported. Immune checkpoint inhibitors targeting programmed cell death receptor 1 and its ligand are the most commonly associated biologics (2). Vedolizumab, a monoclonal antibody that inhibits the gut-selective α4β7 integrin, is administered intravenously to treat ulcerative colitis (UC) (3). While cutaneous adverse reactions like psoriasis and acneiform eruptions have been documented (4, 5), vedolizumab has rarely been linked to autoimmune bullous diseases.
Here, we report a case of bullous pemphigoid (BP) induced by vedolizumab, with immunopathological findings and serological evidence of autoimmunity against BP180, and discuss potential pathogenic mechanisms.
CASE REPORT
A 50-year-old male with a history of UC presented to the dermatology clinic due to erythema and blisters lasting for approximately 2 months. Two months prior, the patient had begun vedolizumab treatment for UC, receiving 300 mg doses administered intravenously at weeks 0, 2 and 6, followed by maintenance infusions every 8 weeks. After the vedolizumab treatments at weeks 0 and 2, the patient developed scattered red papules across his body, which were left untreated. Following vedolizumab treatment at week 6, the patient developed extensive confluent erythema and bullae, without itching or pain. The gastroenterologist prescribed prednisone 35 mg once daily (qd) to treat the skin lesions and advised the patient to visit the dermatology clinic for further evaluation.
Upon physical examination, scattered erythema and tense blisters were found on his trunk and limbs, with some confluent erythema and tense bullae present on the lateral sides of his lower chest and upper abdomen (Fig. 1a). No mucous membrane was involved. A shave biopsy of a newly formed, non-ruptured bulla was sent for routine haematoxylin and eosin staining and revealed a subepidermal blister containing sparse lymphocytes and eosinophils, with moderate perivascular lymphocytic infiltrate and scattered eosinophils in the superficial dermis (Fig. 1b and e). A punch biopsy of perilesional skin was sent for direct immunofluorescence (DIF) and showed positive linear IgG and C3 staining along the basement membrane (BMZ) (Fig. 1c and d), while IgA and IgM were negative.
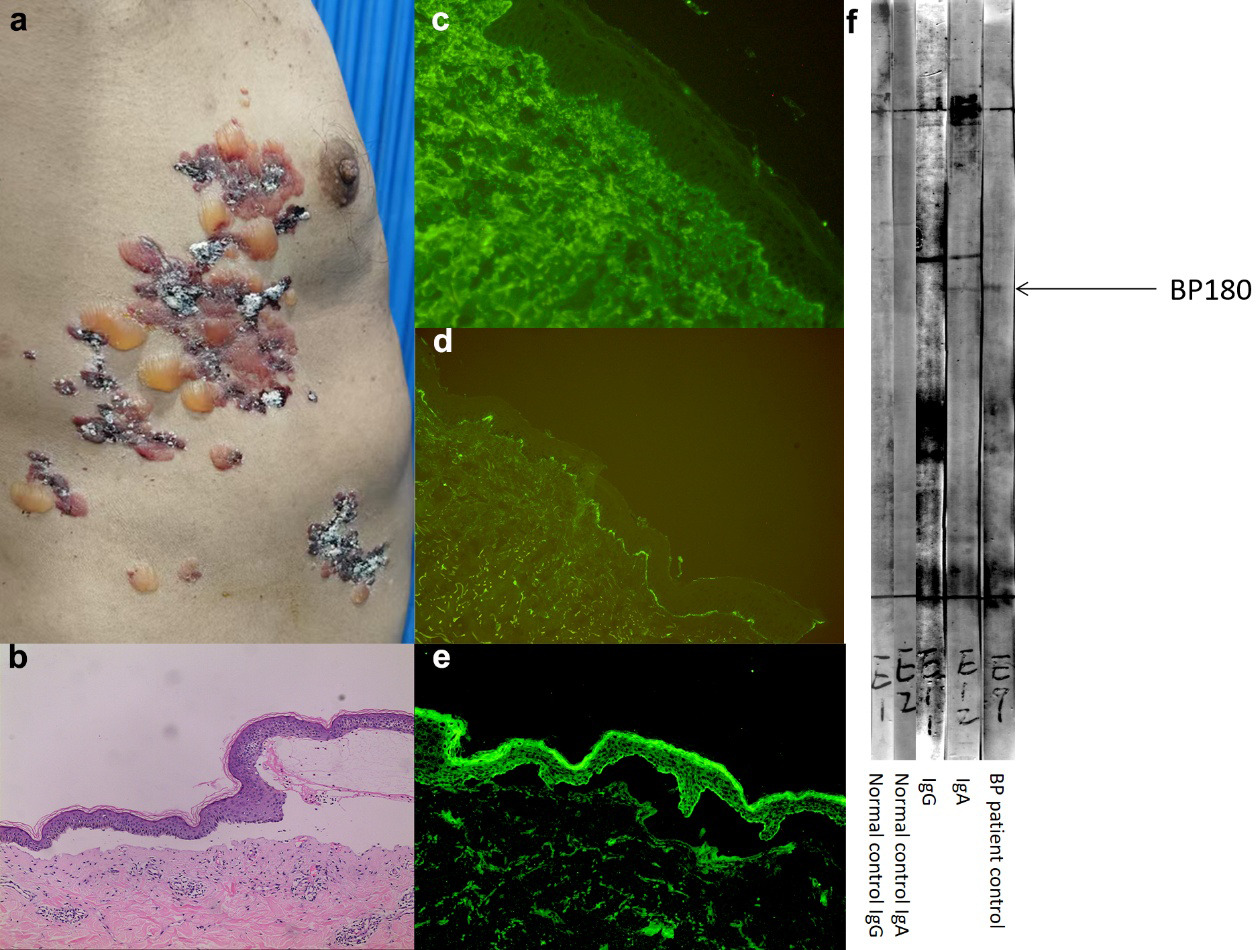
Fig. 1. Clinical and laboratory findings. (a) Scattered haemorrhagic blisters and vesicles on the patient's chest and abdomen. (b) The histopathological examination reveals a subepidermal blister with an inflammatory infiltrate consisting predominantly of eosinophils and lymphocytes. (c) Faint linear deposition of IgG along the basement membrane (BMZ) by direct immunofluorescence (DIF). (d) Linear deposition of C3 along the BMZ by DIF. (e) High-magnification (×400) view showing eosinophils and lymphocytes within the dermal infiltrate. (f) Immunoblot (serum dilution 1:10) analysis using epidermal extract revealed a strong IgA reactivity and a weaker IgG reactivity against the 180 kDa antigen (BP180). A lane with serum from a known bullous pemphigoid (BP) patient serves as a positive control for IgG. Immunoblotting with serum from a healthy normal control was negative. (g) Indirect immunofluorescence (serum dilution 1:10) on 1M NaCl-split skin revealing IgG reactivity on the epidermal side of the split.
Serological testing was performed to confirm the diagnosis. While IgG Enzyme-linked immunosorbent assay (ELISA) for the BP180 NC16a domain and BP230 were negative, salt-split skin indirect immunofluorescence (ss-IIF) (serum dilution 1:10) revealed clear IgG reactivity on the epidermal side of the split, a classic finding for BP (Fig. 1e). A parallel ss-IIF assay for IgA autoantibodies was also performed and was negative. Further analysis by immunoblotting (serum dilution 1:10) using normal human epidermal extract revealed serum IgA autoantibodies targeting BP180, accompanied by a weaker but detectable IgG reactivity against the same antigen. Immunoblotting with serum from a healthy normal control was negative (Fig. 1f).
Based on the temporal relationship with drug initiation, clinical presentation and immunopathological findings, a diagnosis of vedolizumab-induced pemphigoid was made. The patient was instructed to permanently discontinue vedolizumab while continuing prednisone 35 mg daily. At a 4-week follow-up, the bullae had resolved completely, leaving residual post-inflammatory erythema.
DISCUSSION
We describe a case of pemphigoid induced by vedolizumab. While rare, this association is supported by a recently published report identifying a similar adverse event (6). The diagnosis is supported by a clear temporal onset, characteristic clinical and histological features, positive DIF, definitive serological evidence and resolution upon drug withdrawal.
A key immunopathological finding in this case was the detection of a dual IgA and IgG autoimmune response against BP180 via immunoblotting. While standard ELISA was negative, the more sensitive immunoblot confirmed the diagnosis and revealed an unusual, IgA-predominant profile. A diagnosis of classic linear IgA bullous dermatosis (LABD) was ruled out by the negative IgA deposition on DIF. The specificity of the immunoblot was confirmed by a negative result from a healthy control.
The weaker IgG signal is likely attributable to the patient’s initiation of systemic prednisone prior to serological analysis, a factor known to suppress antibody titres. Clinical studies have shown that prednisone reduces serum IgG levels more substantially than IgA – for example, a short course of prednisone (mean 16.8 mg/day for 15 days) decreased serum IgG by 22%, compared with only 10% for IgA, while IgM remained unchanged (7). This established differential effect provides a strong precedent for our findings, suggesting that prednisone suppressed the pathogenic IgG response more profoundly than the co-existing IgA response, resulting in the IgA-dominant profile we observed.
The presence of both IgA and IgG autoantibodies targeting BP180 is uncommon in idiopathic pemphigoid and strongly suggests a drug-induced aetiology. This dual-isotype response likely reflects a broad, drug-triggered immune dysregulation resulting in polyclonal B-cell activation against the BP180 antigen. This case, therefore, represents a unique variant of drug-induced pemphigoid, rather than a classic presentation of either BP or LABD.
The mechanism linking a gut-selective integrin inhibitor to this specific autoimmune response remains unclear, but two main hypotheses can be considered. The most established explanation involves immune dysregulation. By blocking the homing of α4β7-expressing lymphocytes to the gut, vedolizumab alters systemic T-cell trafficking (8). This may redirect certain T-cell populations to the skin, disrupting local immune homeostasis and unmasking a latent autoimmune predisposition. This type of paradoxical inflammation is a known phenomenon with other biologics (5).
A second, more speculative hypothesis involves molecular mimicry and epitope spreading. The α4 integrin subunit targeted by vedolizumab shares structural homology with the α6 integrin subunit, a key component of the hemidesmosome in the skin’s BMZ (9). It is theoretically possible that an immune response initiated against α4 integrin could cross-react with α6β4 integrin in the skin. As α6β4 is physically complexed with BP180, such a misdirected attack could lead to collateral damage and exposure of BP180 neoantigens, ultimately triggering the production of the anti-BP180 antibodies we detected (10). We acknowledge this mechanism is speculative; if common, a higher incidence of pemphigoid would be expected, and definitive proof would require complex assays not performed here.
Regardless of the initial trigger, the result was a pathogenic autoimmune response against BP180. Prior reports of cutaneous adverse events from vedolizumab include inflammatory conditions and rare cases of drug-induced lupus (11, 12, 13). Our case expands this adverse event profile to include a severe autoimmune blistering disease.
In conclusion, this case establishes pemphigoid as a potential, albeit rare, adverse reaction to vedolizumab. Clinicians should maintain a high index of suspicion for autoimmune skin events in patients receiving the therapy. If pemphigoid is suspected and initial serology is negative, immunoblotting should be considered.
ACKNOWLEDGEMENTS
We would like to thank Dr. Li Xiaoguang and Dr. Qian Hua for helping us establish the serum immunoblotting diagnosis for AIBD.
REFERENCES
- Bağcı IS, Horváth ON, Ruzicka T, Sárdy M. Bullous pemphigoid. Autoimmun Rev 2017; 16: 445–455. https://doi.org/10.1016/j.autrev.2017.03.010
- Verheyden M, Murrell D, Bilgic A. 12871 A systematic review of drug-induced pemphigoid. J Am Acad Dermatol 2020; 83: AB113. https://doi.org/10.1016/j.jaad.2020.06.539
- Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2013; 369: 699–710. https://doi.org/10.1056/NEJMoa1215734
- Magdaleno-Tapial J, Ferrer-Guillén B, Valenzuela-Oñate C, Esteve-Martínez A. Acneiform eruption induced by vedolizumab. Dermatol Online J 2018; 24: 13030/qt0vg996xr.
- Sody E, Körber A. Psoriasis Induced by Vedolizumab. Inflamm Bowel Dis 2017; 23: E9–E11. https://doi.org/10.1097/MIB.0000000000001011
- Odom MB, Chen JN, Echavarria JF, Gilson RT. A case of bullous pemphigoid in an ulcerative colitis patient treated with vedolizumab. JAAD Case Rep 2025; 66: 176–179. https://doi.org/10.1016/j.jdcr.2025.09.033
- Settipane GA, Pudupakkam RK, McGowan JH. Corticosteroid effect on immunoglobulins. J Allergy Clin Immunol 1978; 62: 162–166. https://doi.org/10.1016/0091-6749(78)90101-x
- Gorfu G, Rivera-Nieves J, Ley K. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr Mol Med 2009; 9: 836–850. https://doi.org/10.2174/156652409789105525
- Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res 2010; 339: 269–280. https://doi.org/10.1007/s00441-009-0834-6
- Aho S, Uitto J. Direct interaction between the intracellular domains of bullous pemphigoid antigen 2 (BP180) and beta 4 integrin, hemidesmosomal components of basal keratinocytes. Biochem Biophys Res Commun 1998; 243: 694–699. https://doi.org/10.1006/bbrc.1998.8162
- Sedano R, Dhaliwal I, Ramsewak D, Jairath V. Drug-induced lupus associated with vedolizumab in a patient with Crohn’s disease. Inflamm Bowel Dis 2021; 27: e47–e48. https://doi.org/10.1093/ibd/izaa331
- Licata G, Gambardella A, De Rosa A, Calabrese G, Alfano R, Argenziano G. Hidradenitis suppurativa caused by vedolizumab. Dermatitis 2021; 32: e23–e24. https://doi.org/10.1097/DER.0000000000000610
- Choe SI, Bourgeois J, Nidamanuri S, Rubenzik R. Pyoderma gangrenosum in an ulcerative colitis patient on vedolizumab. Cureus 2024; 16: e69219. https://doi.org/10.7759/cureus.69219