SHORT COMMUNICATION
Concurrent Drug-induced Rowell Syndrome and Acute Generalized Exanthematous Pustulosis
Yu-Han Alice HSU1,2![]() , Kuan-Yu CHU1,2 and Cheng-Che Eric LAN1,2*
, Kuan-Yu CHU1,2 and Cheng-Che Eric LAN1,2*
1Department of Dermatology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan, and 2Department of Dermatology, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan. *Email: laneric@cc.kmu.edu.tw
Citation: Acta Derm Venereol 2026; 106: adv-2026-0400. DOI: https://doi.org/10.2340/actadv.v106.adv-2026-0400.
Copyright: © 2026 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Feb 4, 2026. Accepted after revision: Mar 19, 2026.
Published: Apr 16, 2026.
Competing interests and funding: The authors have no conflicts of interest to declare.
Written informed consent for publication was obtained from the patient. IRB approval was not required for this case report.
Acute generalized exanthematous pustulosis (AGEP) is a severe drug-related pustular eruption characterized by the abrupt onset of sterile nonfollicular pustules. Rowell syndrome (RS) is a rare variant of cutaneous lupus erythematosus presenting with erythema multiforme–like targetoid lesions. Although medications have been associated with both conditions, their concurrent presentation is rare. Here, we report a patient with systemic lupus erythematosus (SLE) who developed AGEP accompanied by the acute onset of typical cutaneous manifestations of RS.
CASE PRESENTATION
A 45-year-old woman presented with a 3-day history of generalized pruritic erythematous rash with a fever of up to 39.5 °C, fatigue and dyspnoea. She had been diagnosed with SLE 2 weeks earlier, manifesting with a malar rash. Serological testing revealed positive antinuclear antibodies (ANA, >1 : 160, speckled pattern), anti-Ro (>240 U/mL), anti-Smith antibodies (14 U/mL; reference range, <7 U/mL) and low complement 4 level (6.7 mg/dL; reference range, 10–40 mg/dL). The patient was initially treated with prednisolone (5 mg/day), hydroxychloroquine 200 mg/day and azathioprine 50 mg/day. Two weeks after treatment initiation, the malar rash persisted. Therefore, the azathioprine dose was increased to 100 mg/day, while hydroxychloroquine was maintained at 200 mg/day. Three days after the azathioprine dose escalation (17 days after initiation of hydroxychloroquine and azathioprine), the patient developed an acute exacerbation of widespread cutaneous eruptions. Physical examination revealed generalized maculopapular eruptions with nonfollicular sterile pustules and erosions on the face and trunk (Fig. 1a, d). Erythematous papules were noted on the dorsal hands, fingers and periungual areas, with sparing of the knuckles, consistent with chilblain lupus (Fig. 1b). In addition, multiple reddish to violaceous annular patches with a targetoid configuration and without scaling were observed on the upper limbs, dorsal hands and shoulders, resembling erythema multiforme (Fig. 1a, b and c). No oral or genital mucosal involvement was present. Laboratory test showed leukocytosis (12,000 /μL) with neutrophilia (80%), and immunoglobulin M antibodies for herpes simplex virus (HSV) and Mycoplasma pneumoniae were negative. Histopathological examination of the pustule of the trunk revealed subcorneal neutrophilic accumulation (Fig. 2a) and perivascular lymphohistiocytic infiltrates with numerous eosinophils (Fig. 2b), consistent with AGEP. A biopsy from a targetoid lesion of the upper limb demonstrated basal vacuolar degeneration with perivascular lymphocytic infiltration (Fig. 2c). Direct immunofluorescence was negative. With a clinical impression of azathioprine and hydroxychloroquine induced AGEP with acute exacerbation of RS, the suspected culprit drugs were discontinued, and intravenous methylprednisolone (40 mg every 8 h) and topical desoximetasone 0.25 % ointment were initiated. Fever and malaise subsided, the pustules gradually dried, and the targetoid eruptions resolved with post-inflammatory hyperpigmentation within two weeks. (Fig. 1e). No recurrence of skin lesions was observed during 1 year of follow-up. The patient remains in remission on prednisolone 5 mg daily and mycophenolic acid 360 mg twice daily.

Fig. 1. Cutaneous manifestations at presentation and follow-up. (a) Erythematous annular patches with a targetoid configuration on the upper limbs. Maculopapular eruptions with pustules and erosions involving the face and trunk. (b) Chilblain lupus and targetoid patches on the dorsal hands. (c) Targetoid patches on shoulder and upper limbs. (d) Nonfollicular centric pustules with erythema on the chest. (e) Resolution of the lesion in 1 week.
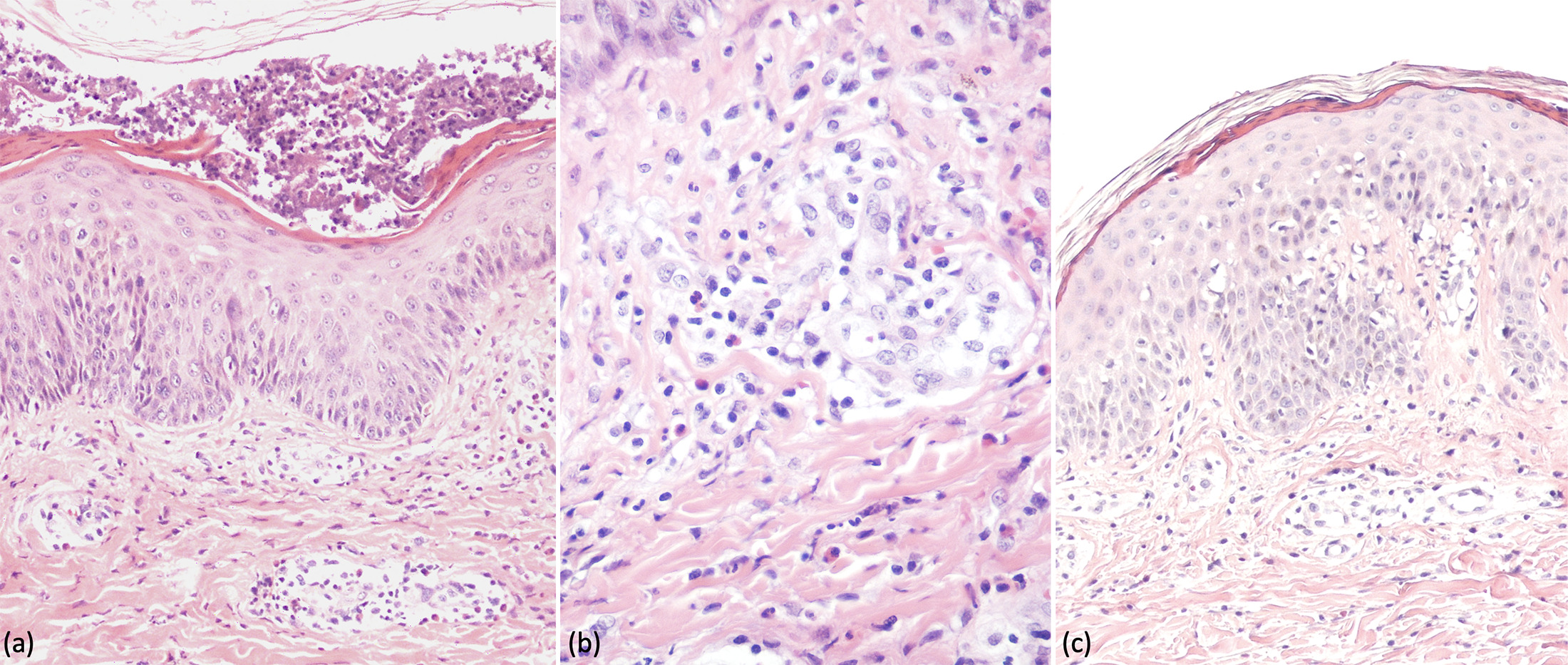
Fig. 2. Histopathologic features. (a,b) Haematoxylin and eosin staining from specimen on the trunk revealed subcorneal neutrophilic accumulation and perivascular infiltration of lymphocytes, histiocyte and numerous eosinophils. (a, magnification ×100) (b, magnification ×200). (c) Haematoxylin and eosin staining from the targetoid lesion on the limb revealed basal vacuolar degeneration with perivascular lymphocytic infiltration (magnification ×100).
DISCUSSION
RS is a rare disorder that usually appears in middle-aged female characterized by the coexistence of lupus erythematosus (LE) and erythema multiforme (EM)-like lesions. Diagnostic criteria included the presence of LE lesions (especially discoid lupus erythematosus (DLE) and/or chilblain), EM-like lesions, positive speckled ANA, anti-Ro/SSA or anti-La/SSB antibodies and rheumatoid factor (1). The absence of trigger factors for EM including infection (e.g. mycoplasma pneumonia, HSV) was also needed for the diagnosis of RS (2). Herold M et al. proposed that RS should be considered a distinct variant within the spectrum of cutaneous LE, based on the enrichment of CD123+plasmacytoid dendritic cells, which aid in its distinction from EM. Furthermore, unlike subacute cutaneous LE (SCLE), annular lesions in RS show a targetoid configuration, with a central dusky erythematous zone surrounded by peripheral erythema (3).
Drug-induced lupus (DIL) is an autoimmune syndrome triggered by certain medications and may manifest as SLE, SCLE, or less commonly chronic cutaneous lupus erythematosus (CCLE), with cutaneous forms often presenting with generalized rash and photosensitivity (2). Procainamide and hydralazine are the most common causes of DIL (4). Drug-associated RS has been reported in association with some medications (Table I). The latency period between drug exposure and eruption varied considerably, ranging from a few hours after re-exposure to several months of treatment. Clinical outcomes were generally favorable, with most patients showing improvement following withdrawal of the offending drug and treatment with systemic corticosteroids or antimalarial agents. Compared with previously reported cases, the present case is unique in that the eruption showed features of AGEP, which has not been previously reported in drug-induced RS.
Table I. Published cases of drug-induced Rowell syndrome
| Author | Patient profile | Cutaneous findings | Culprit drug | Systemic symptoms | Latency | Laboratory data | Treatment | Time of remission after drug withdrawal |
|---|---|---|---|---|---|---|---|---|
| Kacalak-Rzepka et al. (5) | 51F with SLE | Target-like lesions on the face and neck, erosion on buttocks and thighs, mucosal involvement | Sodium valproate | Arthralgia, general fatigue and subclinical fever | 5 months; 2 weeks when rechallenging | ANA+anti-Ro+anti-La+RF + | Prednisolone | 3 weeks |
| Murad et al. (6) | 81F | Targetoid lesions on head, neck, back | Terbinafine | None | 3 weeks | ANA+anti-Ro+ | Systemic corticosteroids | 10 days |
| Pozharashka et al. (7) | 67F with SCLE | Target lesions with haemorrhagic crusts at the margins on neck, chest, upper back and shoulder | Omeprazole | None | >1 year | ANA+anti-Ro+anti-La+ | Hydroxychloroquine, methylprednisolone | 1 month |
| Baroni et al. (8) | ˊ70F | Target lesions on back, chest, extremities and face | Norfloxacin | None | Hours after the third dose (re-exposure) | ANA+anti-Ro+anti-La+RF + | Hydroxychloroquine, prednisone | Not reported |
| Champagne et al. (9) | 65F | Targetoid lesions with central blisters on head, trunk and limbs, mucosal erosions and epidermal detachment | Terbinafine | Fever and tachycardia | 3 weeks | ANA+anti-Ro+ RF+ |
Prednisolone, hydroxychloroquine | Rapid improvement |
| Schissler et al. (10) | 43F, SCLE | Target-like lesions with erosion and crust on chest and arms, oral mucosal involvement | Esomeprazole | None | 6 months | ANA+anti-Ro+ | Prednisolone, hydroxychloroquine | 1 year |
| Present case | 45F. SLE | Targetoid patches on trunk and upper limbs, pustules on trunk, erosions on face | Hydroxychloroquine, azathioprine | Fever, fatigue and dyspnoea | 17 days | ANA+anti-Ro+anti-La+ | Methylprednisolone | 2 weeks |
|
ANA+:Antinuclear antibodies positive; anti-La+:Anti-La/SSB antibodies positive; anti-Ro+:Anti-Ro/SSA antibodies positive; F:female; RF+:Rheumatoid factor positive; SCLE:subacute cutaneous lupus erythematosus; SLE:systemic lupus erythematosus. |
||||||||
AGEP is a drug eruption characterized by the abrupt onset of numerous, small, nonfollicular sterile pustules on an erythematous base, typically accompanied by fever >38 °C and leukocytosis, and resolving rapidly following drug discontinuation in 15 days. In the present case, the diagnosis was supported by a EuroSCAR score of 7, consistent with probable AGEP. AGEP is commonly associated with pristinamycin, aminopenicillins, quinolones, antimalarials, sulfonamides, terbinafine, and diltiazem. (11). Azathioprine-induced AGEP has been reported in a few articles (12, 13, 14). In this case, the eruption occurred 17 days after initiation of hydroxychloroquine and azathioprine and three days after azathioprine dose escalation, suggesting a drug-induced reaction. Although the short interval following azathioprine dose escalation may indicate a potential role for azathioprine, hydroxychloroquine is also a recognized trigger of AGEP, therefore, both drugs remain possible culprits.
Histopathologically, RS demonstrates interface dermatitis with basal vacuolar change, and AGEP demonstrates subcorneal or intraepidermal pustules, papillary dermal edema and a dense mixed perivascular infiltrate rich in neutrophils and eosinophils.
AGEP and DIL share drug-specific T-cell–mediated mechanisms. In AGEP, drug-specific T cells activate innate immune pathways, leading to the release of interleukin (IL)-1, IL-17 and tumour necrosis factor-α, which promote neutrophilic infiltration. In DIL, T-cell–mediated immune dysregulation results in autoantibody production and immune complex deposition (15). These observations suggest that drug-induced T-cell activation may trigger distinct immune pathways, giving rise to divergent clinical manifestations that can occur simultaneously in the same individual.
In conclusion, we report a rare case of AGEP with concurrent Rowell syndrome in lupus erythematosus, highlighting that early recognition and drug withdrawal result in favorable outcomes.
REFERENCES
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol 2012; 67: 417–421. https://doi.org/10.1016/j.jaad.2011.10.012
- Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatologic aspects. Lupus 2009; 18: 935–940. https://doi.org/10.1177/0961203309106176
- Herold M, Nielson CB, Braswell D, Merkel K, Walker A, Auerbach J, et al. Clinicopathologic comparison of Rowell syndrome, erythema multiforme, and subacute cutaneous lupus erythematosus. J Am Acad Dermatol 2019; 81: 1435–1438. https://doi.org/10.1016/j.jaad.2019.06.008
- Siegel CH, Sammaritano LR. Systemic lupus erythematosus: a review. JAMA 2024; 331: 1480–1491. https://doi.org/10.1001/jama.2024.2315
- Kacalak-Rzepka A, Kiedrowicz M, Bielecka-Grzela S, Ratajczak-Stefanska V, Maleszka R, Mikulska D. Rowell’s syndrome in the course of treatment with sodium valproate: a case report and review of the literature data. Clin Exp Dermatol 2009; 34: 702–704. https://doi.org/10.1111/j.1365-2230.2008.02972.x
- Murad A, Shudell E, Mulligan N. Rowell’s syndrome induced by terbinafine. BMJ Case Rep 2015; 2015: bcr2015210360. https://doi.org/10.1136/bcr-2015-210360
- Pozharashka J, Dourmishev L, Balabanova M, Vassileva S, Miteva L. Rowell’s syndrome triggered by omeprazole. Acta Dermatovenerol Croat 2019; 27: 124–126.
- Baroni A, Piccolo V, Russo T, Cozzolino D, Mascolo M, Chessa MA. Norfloxacin-induced subacute cutaneous lupus with erythema multiforme-like lesions: the enigma of the Rowell syndrome. J Dtsch Dermatol Ges 2014; 12: 1039–1042. https://doi.org/10.1111/ddg.12392
- Champagne C, Ratnavel R, Wong T. Severe Rowell syndrome associated with oral terbinafine. Clin Exp Dermatol 2012; 37: 822–823. https://doi.org/10.1111/j.1365-2230.2012.04363.x
- Schissler C, Banea S, Tortel MC, Mahé A. Un nouveau cas de syndrome de Rowell. Ann Dermatol Venereol 2017; 144: 263–267. https://doi.org/10.1016/j.annder.2017.02.005
- Sidoroff A, Dunant A, Viboud C, Halevy S, Bavinck JNB, Naldi L, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR). Br J Dermatol 2007; 157: 989–996. https://doi.org/10.1111/j.1365-2133.2007.08156.x
- Elston GE, Johnston GA, Mortimer NJ, Harman KE. Acute generalized exanthematous pustulosis associated with azathioprine hypersensitivity. Clin Exp Dermatol 2007; 32: 52–53. https://doi.org/10.1111/j.1365-2230.2006.02257.x
- Parisi R, Shah H, Navarini AA, Muehleisen B, Ziv M, Shear NH, et al. Acute generalized exanthematous pustulosis: clinical features, differential diagnosis, and management. Am J Clin Dermatol 2023; 24: 557–575. https://doi.org/10.1007/s40257-023-00779-3
- Avgerinos A, Giannikaki E, Krüger-Krasagakis S, Koutroubakis IE. Acute generalized exanthematous pustulosis induced by azathioprine in a patient with ulcerative colitis. Am J Gastroenterol 2011; 106: 1005–1007. https://doi.org/10.1038/ajg.2010.500
- Chang C, Gershwin ME. Drugs and autoimmunity--a contemporary review and mechanistic approach. J Autoimmun 2010; 34: J266–75. https://doi.org/10.1016/j.jaut.2009.11.012