ORIGINAL REPORT
Efficacy and Safety of Abrocitinib for Moderate-to-severe Atopic Dermatitis in Adults: A 52-week Real-world Multicentre Study- IL-AD (Italian Landscape Atopic Dermatitis)
Elena PEZZOLO1*![]() , Mariateresa ROSSI2
, Mariateresa ROSSI2![]() , Raul Armando LUYO SABOYA1
, Raul Armando LUYO SABOYA1![]() , Piergiorgio MALAGOLI3
, Piergiorgio MALAGOLI3![]() , Alessandra NARCISI4,5
, Alessandra NARCISI4,5![]() , Silvia M. FERRUCCI6
, Silvia M. FERRUCCI6![]() , Giampiero GIROLOMONI7
, Giampiero GIROLOMONI7![]() , Anna BALATO8
, Anna BALATO8![]() , Flavia MANZO MARGIOTTA9,10
, Flavia MANZO MARGIOTTA9,10![]() , Claudio SCIARRONE11, Caterina FOTI12
, Claudio SCIARRONE11, Caterina FOTI12![]() , Massimo GOLA13
, Massimo GOLA13![]() , Maria ESPOSITO14
, Maria ESPOSITO14![]() , Maddalena NAPOLITANO15
, Maddalena NAPOLITANO15![]() , Carlotta GURIOLI16
, Carlotta GURIOLI16![]() , Michela ORTONCELLI17
, Michela ORTONCELLI17![]() , Francesca SATOLLI18
, Francesca SATOLLI18![]() , Maria L. MUSUMECI19
, Maria L. MUSUMECI19![]() , Paola SAVOIA20
, Paola SAVOIA20![]() , Fabrizio AMORUSO21, Anna G. BURRONI22
, Fabrizio AMORUSO21, Anna G. BURRONI22![]() , Cristina Beatrice SPIGARIOLO23
, Cristina Beatrice SPIGARIOLO23![]() , Piergiacomo CALZAVARA-PINTON2
, Piergiacomo CALZAVARA-PINTON2![]() , Antonio COSTANZO4,5
, Antonio COSTANZO4,5![]() , Luigi GARGIULO4,5
, Luigi GARGIULO4,5![]() , Francesca BAREI6
, Francesca BAREI6![]() , Martina MAURELLI7
, Martina MAURELLI7![]() , Marco ROMANELLI9
, Marco ROMANELLI9![]() , Federica VERONESE24
, Federica VERONESE24![]() , Benedetta TIRONE12
, Benedetta TIRONE12![]() , Maria C. FARGNOLI25
, Maria C. FARGNOLI25![]() , Cataldo PATRUNO24
, Cataldo PATRUNO24![]() , Simone RIBERO17
, Simone RIBERO17![]() , Marco VACCINO19 and Luigi NALDI1
, Marco VACCINO19 and Luigi NALDI1![]()
1Department of Dermatology, San Bortolo Hospital, Vicenza, Italy, 2Dermatology Department, University of Brescia, ASST Spedali Civili of Brescia, Brescia, Italy, 3Department of Dermatology, Dermatology Unit Azienda Ospedaliera San Donato Milanese, Milan, Italy, 4Dermatology Unit, IRCCS Humanitas Research Hospital, Rozzano, MI, Italy, 5Department of Biomedical Sciences, Humanitas University, Pieve Emanuele, Italy, 6Dermatology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy, 7Section of Dermatology and Venereology, Department of Medicine, University of Verona, Verona, Italy, 8Dermatology Unit, University of Campania L. Vanvitelli, Naples, Italy, 9Department of Dermatology, University of Pisa, Pisa, Italy, 10Health Sciences Interdisciplinary Center, Sant’Anna School of Advanced Studies, Pisa, Italy, 11Dermatology Clinic, Department of Health Science, University of Eastern Piedmont, Novara, Italy, 12Unit of Dermatology, Department of Precision and Regenerative Medicine and Ionian Area, University of Bari Aldo Moro, Bari, Italy, 13Allergological and Pediatric Dermatology Unit, Department of Health Sciences, University of Florence, Florence, Italy, 14Unit of Dermatology, Department of Biotechnological and Applied Clinical Sciences, University of L'Aquila, L'Aquila, Italy, 15Section of Dermatology, Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy, 16Dermatology Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Policlinico S. Orsola-Malpighi, Bologna, Italy, 17Dermatologic Clinic, Department of Medical Science, University of Turin, Turin, Italy, 18Dermatology Unit, Department of Medicine and Surgery, University of Parma, Parma, Italy, 19Dermatology Clinic, University of Catania, Catania, Italy, 20Dermatology Unit, Department of Health Sciences (DiSS), School of Medicine, Università del Piemonte Orientale (UPO), Novara, Italy, 21Dermatology Unit, Azienda Ospedaliera di Cosenza, Cosenza, Italy, 22Department of Dermatology, Dipartimento di Scienze della Salute (DISSAL), University of Genoa, IRCC S Ospedale Policlinico San Martino, Genoa, Italy, 23Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Milan, Italy, 24Dermatology Unit, AOU Maggiore della carità Hospital, Novara, Italy, and 25Department of Health Sciences, University Magna Graecia of Catanzaro, Catanzaro, Italy
Corr: Elena Pezzolo, Dermatology Unit, San Bortolo Hospital, Viale Ferdinando Rodolfi, 37, 36100, Vicenza, Italy. *Email: elena.pezzolo@gmail.com
Key words: Abrocitinib; Atopic Dermatitis; Efficacy; Itch; Minimal Disease Activity; Safety.
Citation: Acta Derm Venereol 2026; 106: adv-2025-0193. DOI: https://doi.org/10.2340/actadv.v106.adv-2025-0193.
Copyright: © 2026 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Nov 16, 2025. Accepted after revision: Mar 17, 2026.
Published: Apr 14, 2026.
Competing interests and funding: The authors have no conflicts of interest to declare.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abrocitinib, an oral selective Janus kinase 1 inhibitor, is approved for the treatment of moderate-to-severe atopic dermatitis (AD) in adults, but real-world evidence remains limited. We retrospectively collected data from 187 adult patients (mean age: 37.9 years; 60.4% male), with AD and up to one-year exposure to abrocitinib either 100 or 200 mg, between 2023 and 2025 across 22 Italian dermatology centres. Nearly half of patients were bio-experienced and 80 % had difficult-to-treat areas. Beginning at week 8, with either 100 (59%) or 200 mg, 52 % achieved Investigator Global Assessment 0/1, significantly increasing to 88 % at week 52. Similarly, Eczema Area and Severity Index (EASI) and itch had significantly higher 1-year response: increasing from 27.8% and 34.7%, respectively, at week 8, 79.6 % achieved EASI-90 and 73.5% reached Peak Pruritus-Numerical Rating Scale 0/1. Minimal disease activity, a stringent composite endpoint, was met by 18 % at week 8 and by 66 % at week 52. Safety was good, with mild adverse events mostly including abdominal pain, upper respiratory infections and headache. The 100 mg dose had a significantly better safety profile, with efficacy similar to 200 mg. These data confirm the efficacy and safety of abrocitinib, with early, progressive clinical benefits sustained through 1 year.
Significance
This multicentre real-world study provides novel evidence on the long-term effectiveness and safety of abrocitinib in adults with moderate-to-severe atopic dermatitis (AD). Among 187 patients followed up to one year, abrocitinib induced rapid, progressive improvement across disease signs, symptoms and patients’ quality of life, with almost 90 % of them achieving clear or almost clear skin, more than 70% reaching no or minimal pruritus and two-thirds obtaining minimal disease activity at week 52. The treatment was well tolerated, particularly at the 100 mg dose. This study contributes relevant real-world insights supporting the positioning of abrocitinib as an effective, well-tolerated systemic option for AD.
Atopic dermatitis (AD) is a chronic, relapsing inflammatory skin disease characterized by intense pruritus, erythema and eczematous lesions (1). It imposes a substantial burden on patients due to persistent itching, sleep disturbance and impaired quality of life, often leading to psychological distress and social limitations (2). Given the complex pathogenesis of AD, which involves immune dysregulation, skin barrier dysfunction and environmental triggers, recent advances have focused on targeted therapies addressing specific inflammatory pathways (3, 4). Despite these advances, many patients remain inadequately controlled, underscoring the need for effective and safe long-term therapeutic options (5, 6, 7).
The treatment landscape for moderate-to-severe AD has evolved significantly in recent years with the development of targeted therapies, including monoclonal antibodies (dupilumab, tralokinumab and lebrikizumab) and small molecules (upadacitinib, abrocitinib and baricitinib) (4). These small molecules interfere with key signalling pathways involved in AD pathogenesis by inhibiting cytokines critical to the inflammatory response.
Abrocitinib, an oral selective Janus kinase 1 (JAK1) inhibitor administered once daily, is approved for the treatment of moderate-to-severe AD in adults (4, 8, 9). Multiple clinical trials have demonstrated its efficacy and safety in patients with moderate-to-severe AD compared with both placebo and dupilumab; however, real-world evidence is still needed to evaluate the achievement of optimal treatment targets (10, 11). We previously conducted a 16-week multicentre retrospective study involving 85 adults with moderate-to-severe AD treated with abrocitinib (12). That analysis confirmed the short-term effectiveness and safety of abrocitinib, showing substantial improvements in disease severity, pruritus and sleep quality, with no significant safety concerns.
In the present study, we aimed to assess the long-term efficacy and safety of abrocitinib in a real-world population of adults with AD over a period of up to 1 year.
MATERIALS AND METHODS
Study design
We conducted a retrospective cohort study across 22 Italian tertiary dermatology referral centres.
Eligible participants were adults (≥≥ years), diagnosed with moderate-to-severe AD by a dermatologist, who received abrocitinib (100 mg or 200 mg orally once daily) between 1 June 2023 and 1 June 2025. Treatment eligibility was based on the EuroGuiDerm guidelines for AD, the Summary of Product Characteristics and the criteria established by the Italian Medicines Agency (AIFA), which define the clinical indications for initiating JAK inhibitor therapy in appropriate candidates (1, 4, 5). Specifically, patients were eligible if they had moderate-to-severe AD with Eczema Area and Severity Index (EASI) score ≥24 requiring systemic treatment, or if they had inadequate response, intolerance or contraindications to cyclosporine A, or previous use of biologics for AD treatment with unsatisfactory clinical response or adverse events (AEs). According to AIFA recommendations, either 100 mg or 200 mg abrocitinib could be prescribed at the physician’s discretion. All patients were encouraged to use emollients daily. Topical corticosteroids of different potencies and calcineurin inhibitors were permitted only for short-term management of acute localized flares. No additional systemic therapies for AD were administered during the study period. Follow-up visits were scheduled according to each centre’s timetable and patient availability. To be included, patients were required to have at least 1 follow-up visit after treatment initiation. The follow-up duration was up to 52 weeks.
The study was conducted in accordance with the principles of the Declaration of Helsinki and with applicable national and European regulations on patient data protection, privacy and confidentiality (13, 14). As this was a retrospective, non-interventional analysis of anonymized data obtained from routine clinical practice, formal approval by an institutional ethics committee was not required according to Italian regulations. All patients had provided written informed consent for the use of their anonymized clinical data collected during routine visits. The study complied with local ethical standards and adhered to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines for cohort studies (15).
Data collection
Clinical data were systematically collected and stored in a centralized electronic database accessible to all participating centres. Collected information included demographics (age, sex), medical history (body mass index (BMI), smoking status, atopic and cardiometabolic comorbidities), AD duration and phenotype, AD severity, patient’s bio-experience status (abrocitinib-naïve vs previously treated with a biologic, such as dupilumab, tralokinumab or lebrikizumab), starting dosage of abrocitinib (100 mg or 200 mg), treatment outcomes and AEs.
The population was stratified into two groups according to daily abrocitinib dosage (100 mg or 200 mg).
Disease severity and therapeutic response were assessed using the Eczema Area and Severity Index (EASI), Investigator Global Assessment (IGA), Peak Pruritus-Numerical Rating Scale (PP-NRS) and Sleep Disturbance (SD-NRS) (16). The EASI, ranging from 0 (no disease) to 72 (most severe disease), measures the extent and severity of AD (17). The IGA, a 5-point physician-rated scale from 0 (clear) to 4 (severe), evaluates overall lesion severity (17). The PP-NRS and SD-NRS, ranging from 0 (no symptoms) to 10 (very severe symptoms), assess symptom intensity over the previous 24 h. Data were collected at baseline (treatment initiation) and at 8, 16, 32 and 52 weeks post-baseline.
Efficacy assessment
For EASI, the proportion of patients achieving ≥75%, ≥90% and 100% reduction from baseline (EASI-75, EASI-90, and EASI-100, respectively), as well as absolute scores ≤7 and ≤3 at each time point, were evaluated (16). For IGA, the proportion of patients achieving a score of 0 or 1 (“clear” or “almost clear”) was calculated. Similarly, for PP-NRS and SD-NRS, we assessed the proportions of patients achieving aa4-point reduction from baseline and absolute scores of 0 or 1 (17). Minimal Disease Activity (MDA), a stringent composite endpoint, was defined as the simultaneous achievement of EASI-90 and PP-NRS 0/1 (18, 19). Finally, abrocitinib efficacy was compared between patients receiving 100 mg and those receiving 200 mg daily.
Safety assessment
All AEs occurring during the one-year follow-up were recorded, including severe AEs, those leading to treatment discontinuation and those requiring dose reduction.
Statistical analysis
For the analyses, the number and percentage of patients achieving the outcomes of interest were calculated. Continuous variables were presented as mean and standard deviation (SD) or median and interquartile range (IQR), while categorical variables were reported as absolute numbers and percentages. Percentages were based on the number of participants in the specific population unless otherwise indicated. Analyses were performed separately by time point and treatment group. Continuous variables with a normal distribution were compared using the Student t-test, whereas variables with a non-normal distribution were compared using the Mann–Whitney U test. Categorical outcomes were compared using the Chi-squared test when sample sizes were adequate or Fisher’s exact test for small sample sizes. Proportions of patients achieving clinical outcomes at different time points (Weeks 8, 16, 32 and 52) were compared using Generalized Estimating Equations (GEE) to account for data correlation and handle missing values under the assumption that data were missing at random (20). A two-tailed p-value 0.05 was considered statistically significant. All analyses were performed using IBM SPSS Statistics (version 26, IBM Corporation, Armonk, NY, USA).
RESULTS
Baseline patients’ characteristics
The cohort included 187 patients, 113 (60.4%) male, with a mean (±SD) age of 37.9 (±15.5) years. Baseline characteristics of the study population are summarize in Table I.
Table I. Demographic and clinical characteristics of the study population at baseline
| Characteristics | No. | Abrocitinib 200 mg | Abrocitinib 100 mg | p-value* |
|---|---|---|---|---|
| Total (%) | 187 (100) | 76 (40.6) | 111 (59.4) | |
| Sex (%) | ||||
| Female | 74 (39.6) | 27 (35.5) | 47 (42.3) | 0.35 |
| Male | 113 (60.4) | 49 (64.5) | 64 (57.7) | 0.35 |
| Age, years, mean (SD) | 37.9 (15.5) | 33.8 (9.7) | 39 (14.1) | 0.003 |
| BMI, mean (SD) | 24.3 (3.7) | 24.6 (3.1) | 24.0 (2.8) | 0.17 |
| Bio-experienced (%) | 85 (45.5) | 37 (48.7) | 48 (43.2) | 0.32 |
| Age at AD onset, years, mean (SD) | 20.4 (19.8 | 13.5 (11.9) | 25.1 (19.2) | <0.0001 |
| AD phenotype (%) | ||||
| Flexural | 164 (87.7) | 64 (84.2) | 100 (90.1) | 0.32 |
| Generalized inflammatory | 8 (4.2) | 3 (3.9) | 5 (4.5) | 0.55 |
| Prurigo nodularis | 4 (2.1) | 1 (1.3) | 3 (2.7) | 0.34 |
| Palmar-plantar | 4 (2.1) | 2 (2.6) | 2 (1.8) | 0.57 |
| Nummular eczema | 3 (1.6) | 2 (2.6) | 1 (0.9) | 0.32 |
| Erythroderma | 3 (1.6) | 3 (3.9) | 0 (0) | 0.05 |
| Generalized lichenoid | 1 (0.5) | 1 (1.3) | 0 (0) | 0.06 |
| Involvement of sensitive areas (%) | 151 (80.7) | 68 (89.5) | 83 (74.8) | 0.01 |
| Face/neck | 122 (65.2) | 62 (81.6) | 60 (54.1) | 0.0001 |
| Hands | 95 (50.8) | 48 (63.2) | 47(42.3) | 0.005 |
| Genitalia | 9 (4.8) | 6 (7.9) | 3 (2.7) | 0.10 |
| Atopic comorbidities (%) | 83 (44.4) | 36 (47.4) | 47(42.3) | 0.49 |
| Allergic rhinitis | 70 (37.6) | 33 (43.4) | 37 (33.3) | 0.16 |
| Allergic conjunctivitis | 47 (25.3) | 24 (31.6) | 23 (20.7) | 0.09 |
| Allergic asthma | 34 (18.2) | 15 (19.7) | 19 (17.1) | 0.65 |
| Food allergy | 17 (9.1) | 7 (9.2) | 10 (9.0) | 0.96 |
| Nasal polyposis | 1 (0.5) | 0 (0) | 1 (0.9) | 0.41 |
| Cardiometabolic comorbidities (%) | 33 (17.9) | 9 (11.8) | 24 (21.6) | 0.06 |
| Arterial hypertension | 16 (8.7) | 3 (3.9) | 13 (11.7) | 0.06 |
| Dyslipidaemia | 15 (8.1) | 3 (3.9) | 12 (10.8) | 0.10 |
| Obesity | 10 (5.4) | 6 (7.9) | 4 (3.6) | 0.37 |
| Cardiovascular disease | 6 (3.2) | 0 (0) | 6 (5.4) | 0.01 |
| Type 2 diabetes | 5 (2.7) | 0 (0) | 5 (4.5) | 0.04 |
| Smoke (%) | 4 (2.1) | 1 (1.3) | 3 (2.7) | 0.34 |
| Other comorbidities (%) | ||||
| Psoriasis | 1 (0.3) | 0 (0) | 1 (0.9) | 0.32 |
| IGA=4 at baseline (%) | 79 (42.2) | 29 (38.2) | 50 (45.0) | 0.31 |
| EASI at baseline, mean (SD) | 20.8 (10.0) | 19.4 (7.7) | 21.8 (7.1) | 0.04 |
| PP-NRS score at baseline, mean (SD) | 7.2 (2.2) | 7.4 (1.9) | 7.2 (1.4) | 0.43 |
| SD-NRS score at baseline, mean (SD) | 5.9 (2.9) | 6.1 (2.5) | 5.8 (2.3) | 0.42 |
|
*Two-tailed Fisher’s exact test for comparison between the categorical variables of two independent groups and Two-tailed Student t-test for comparison between the means of two independent groups. AD:Atopic dermatitis; BMI:Body Mass Index; EASI:Eczema Area and Severity Index; IGA:Investigator Global Asaessment; PP-NRS:Peak Pruritus-Numerica Rating Scale; SD:Standard Deviation; SD-NRS:Sleep Disturbances-NRS. |
||||
Most patients (60%) started on abrocitinib 100 mg, while the remainder received 200 mg. Nearly half of the cohort was bio-experienced, with a higher proportion previously treated with dupilumab (n=64; 75.3%) compared to tralokinumab (n=18; 21.2%) and lebrikizumab (nn3; 3.5%). At baseline, 42.2 % of patients had an IGA of 4, and mean (±SD) scores of EASI, PP-NRS and SD-NRS were 20.8 (±10.4), 7.2 (±2.2) and 5.9 (±2.9), respectively. The most common AD phenotype was flexural (87.7%), with a mean (±SD) age at AD onset of 20.4 (±9.8) years. More than 80 % of patients had involvement of difficult-to-treat areas, including face/neck (65.2%), hands (50.8%) and genitalia (4.8%). Atopic comorbidities were present in 44 % of patients, mainly allergic rhinitis (37.6%), conjunctivitis (25.3%) and asthma (18.2%). Cardiometabolic conditions, including hypertension, dyslipidaemia, obesity, cardiovascular disease and type 2 diabetes, affected nearly 20 % of patients with 2% reporting smoking.
The two treatment groups – abrocitinib 200 mg (n=76, 41%) and 100 mg (n=111, 59%) – were comparable for most demographic and clinical variables. However, patients in the 200 mg group were significantly younger (33.8 ± 9.7 vs 39.0 ± 14.1 years; p=0.003) and had an earlier age at AD onset (13.5 ± 11.9 vs 25.1 ± 19.2 years; p<0.0001) compared to the 100 mg group. Significant differences were also observed in the involvement of sensitive areas: the 200 mg group had a higher prevalence of face/neck (81.6% vs 54.1%; p=0.0001) and hands (63.2% vs 42.3%; p=0.005) involvement compared to the 100 mg group. The mean baseline EASI score was significantly lower in the 200 mg group (19.4 ± 7.7) than in the 100 mg group (21.8 ± 7.1; p=0.04). No statistically significant differences were observed between groups for IGA=4, PP-NRS or SD-NRS scores.
Efficacy outcomes
Efficacy data were pooled for the 100 mg and 200 mg dosages. Beginning at week 8, 55.8 % of patients (n=58) achieved EASI-75, 27.8 % (n=29) reached EASI-90 and 20.2 % (n=21) reached EASI-100, increasing significantly to 96.3 % (n=52), 79.6 % (n=43) and 59.3 % (n=32) by week 52, respectively (p<0.001) (Table SI). Similarly, an absolute EASI ≤7 was achieved by 70.2 % (n=73) and EASI ≤3 by 37.5 % (n=39) at week 8, significantly rising to 98.1 % (n=53) and 94.4 % (n=51) at week 52, respectively (p<0.001). More than half of patients (51.9%; n=54) achieved IGA 0 or 1 at week 8, significantly increasing to 88.9 % (n=48) at week 52, (p<0.001) (Table SI).
Improvements were also observed in pruritus and sleep scores. At week 8, a ≥4-point reduction in PP-NRS and SD-NRS was reported in 70.4 % (n=69) and 62.2 % (n=61) of patients, significantly rising to 86.8 % (n=46; p=0.010) and to 79.2 % (n=42; p=0.024) by week 52, respectively. Absolute PP-NRS 0/1 and SD-NRS 0/1 were achieved by 34.7 % (n=34) and 62.2 % (n=61) of patients at week 8, significantly increasing to 73.5 % (n=39) and 98.1 % (n=52) at week 52, respectively (p<0.001).
Achievement of optimal therapeutic targets at weeks 8, 16, 32 and 52 is shown in Fig. 1.
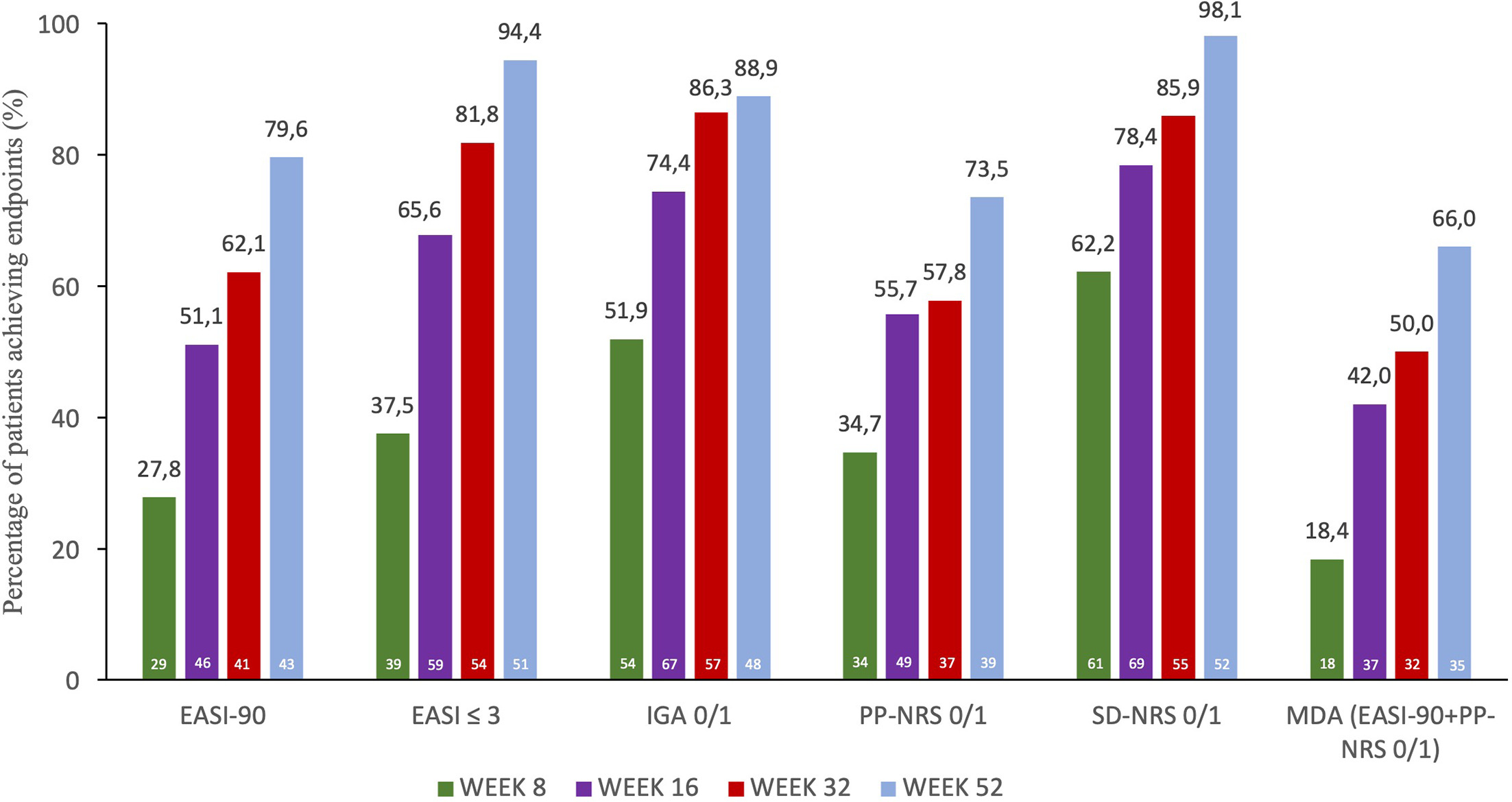
Fig. 1. Efficacy of abrocitinib on achievement of optimal therapeutic targets at weeks 8, 16, 32 and 52. EASI: Eczema Area and Severity Index; IGA: Investigator Global Assessment; MDA: Minimal Disease Activity; PP-NRS: Peak Pruritus-Numerical Rating Scale (NRS); SD-NRS: Sleep Disturbance-NRS. Numbers at the bottom of each bar represent n. Patients number for clinician-reported outcomes was N=104 at week 8, N=90 at week 16, N=66 at week 32 and N=54 at week 52. Patients number for patient-reported outcomes and for MDA was N=98 at week 8, N=88 at week 16, N=64 at week 32 and N=53 at week 52.
Importantly, MDA, a stringent composite endpoint defined as the simultaneous achievement of EASI-90 and PP-NRS 0/1, was reached by 18.4 % of patients (n=18) at week 8, significantly increasing to 42.0 % (n=37; p<0.001) at week 16, 50.0 % (n=32; p<0.001) at week 32 and 66.0 % (n=35; p<0.001) at week 52, respectively (Fig. 2) (18).
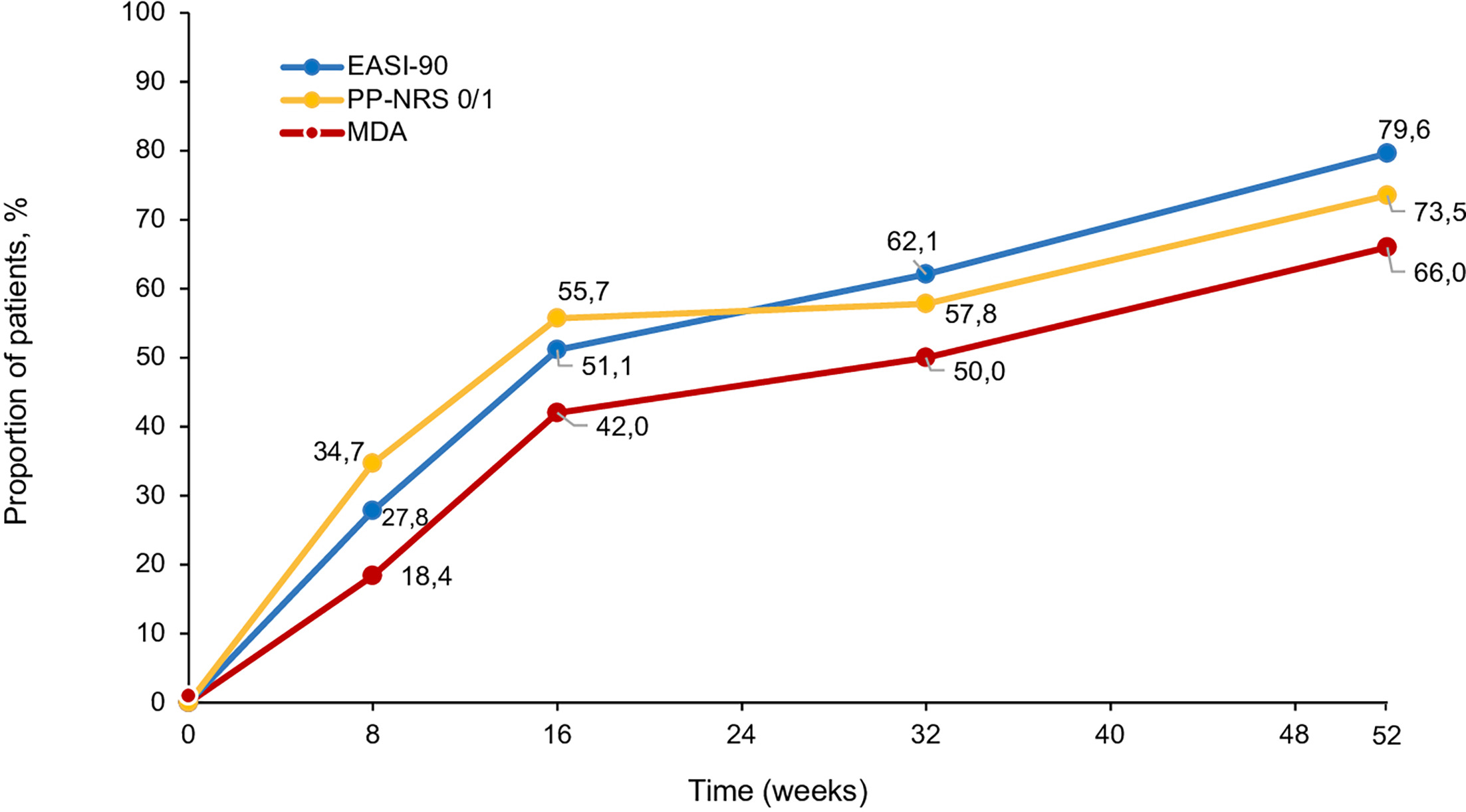
Fig. 2. Achievement of EASI-90, PP-NRS 0/1 and of MDA through week 52. EASI: Eczema Area and Severity Index; MDA: Minimal Disease Activity; PP-NRS: Peak Pruritus Numerical Rating Scale (NRS); SD-NRS: Sleep Disturbance-NRS.
Finally, efficacy outcomes were compared between patients starting on abrocitinib 100 mg versus 200 mg daily. Results stratified by dosage are presented in Table SI. No significant differences in efficacy were observed between the two groups at any time point. An additional subgroup analysis evaluating treatment efficacy according to sex was performed (Table II). No statistically significant differences were observed between male and female patients across the evaluated clinical outcomes at any time point.
Table II. Efficacy outcomes of abrocitinib according to sex at different timepoints throughout the study
| Outcomes | Total patients % W8 |
Males % W8 |
Females % W8 |
p-value* | Total patients % W16 |
Males % W16 |
Females % W16 |
p-value* | Total patients % W32 |
Males % W32 |
Females % W32 |
p-value* | Total patients % W52 |
Males % W52 |
Females % W52 |
p-value* |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EASI 75 | 55.8 | 54.3 | 58.8 | 0.66 | 77.8 | 75.0 | 83.3 | 0.40 | 89.4 | 88.1 | 91.7 | 0.71 | 96.3 | 96.7 | 95.8 | 0.86 |
| EASI 90 | 27.8 | 25.7 | 32.3 | 0.47 | 51.1 | 46.7 | 60.0 | 0.23 | 62.1 | 54.8 | 75.0 | 0.11 | 79.6 | 76.7 | 83.3 | 0.54 |
| EASI 100 | 20.2 | 18.6 | 23.5 | 0.56 | 34.4 | 33.3 | 36.7 | 0.75 | 37.9 | 33.3 | 45.8 | 0.32 | 59.3 | 63.3 | 54.2 | 0.49 |
| EASI≤7 | 70.2 | 71.4 | 67.6 | 0.68 | 84.4 | 81.7 | 90.0 | 0.34 | 93.9 | 92.9 | 95.8 | 0.64 | 98.1 | 100.0 | 91.7 | 0.19 |
| EASI≤3 | 37.5 | 34.3 | 44.1 | 0.32 | 65.6 | 61.7 | 73.3 | 0.28 | 81.8 | 80.9 | 83.3 | 0.80 | 94.4 | 96.7 | 91.7 | 0.39 |
| IGA 0/1 | 51.9 | 51.4 | 52.9 | 0.88 | 74.4 | 71.7 | 80.0 | 0.40 | 86.3 | 83.3 | 91.7 | 0.35 | 88.9 | 93.3 | 83.3 | 0.24 |
| ΔPP-NRS≥4 | 70.4 | 71.2 | 68.7 | 0.81 | 73.8 | 77.6 | 66.7 | 0.27 | 82.8 | 82.5 | 83.3 | 0.93 | 86.8 | 86.2 | 87.5 | 0.89 |
| ΔSD-NRS≥4 | 62.2 | 60.6 | 65.6 | 0.64 | 71.5 | 70.7 | 73.3 | 0.80 | 75.0 | 72.5 | 79.2 | 0.55 | 79.2 | 82.8 | 75.0 | 0.48 |
| PP-NRS 0/1 | 34.7 | 31.8 | 40.6 | 0.39 | 55.7 | 56.9 | 53.3 | 0.75 | 57.8 | 52.5 | 66.7 | 0.26 | 73.5 | 75.9 | 70.8 | 0.67 |
| SD-NRS 0/1 | 62.2 | 59.1 | 68.7 | 0.35 | 78.4 | 75.9 | 83.3 | 0.43 | 85.9 | 85.0 | 87.5 | 0.77 | 98.1 | 96.5 | 100.0 | 0.35 |
| MDA | 18.4 | 16.7 | 21.9 | 0.53 | 42.0 | 44.8 | 36.7 | 0.47 | 50.0 | 45.0 | 58.3 | 0.29 | 66.0 | 65.5 | 66.7 | 0.92 |
|
*Two-tailed Fisher’s exact test. Patients number for clinician-reported outcomes was N=104 (70 males and 34 females) at week 8, N=90 (60 males and 30 females) at week 16, N=66 (42 males and 24 females) at week 32 and N=54 (30 males and 24 females) at week 52. Patients number for patient-reported outcomes was N=98 (66 males and 32 females) at week 8, N=88 (58 males and 30 females) at week 16, N= 64 ( 40 males and 24 females) at week 32 and N=53 (29 males and 24 females) at week 52. EASI:Eczema Area and Severity Index; IGA:Investigator Global Assessment; MDA:Minimal Disease Activity; PP-NRS:Peak Pruritus-Numerical Rating Scale (NRS); SD-NRS:Sleep Disturbance-NRS; W:Week; Δ:delta. |
||||||||||||||||
Safety outcomes
During the study period, 121 (64.7%) patients were evaluable for safety (Table III). AEs occurred in 21.5 % of patients (n=26), with a significantly higher frequency in the abrocitinib 200 mg group (37.8%; n=14) compared with the 100 mg group (16.7%; n=14; p=0.01). The most common AEs were abdominal pain (n=7; 5.8%), upper respiratory tract infections and headache (n=4; 3.3% each). Less frequent AEs included JAK inhibitor-associated acne (n=3; 2.5%), nausea, weight gain and hypercholesterolaemia (n=2; 1.7% each); and herpes simplex, elevated liver enzymes, urinary tract infection and elevated CPK (n=1; 0.8% each). All AEs were mild, with no serious or life-threatening events reported. Two patients (1.6%) discontinued abrocitinib due to AEs, while no dose reductions were required.
Table III. Safety of abrocitinib during the study period
| Totala N=121 |
Abrocitinib 200 mg N=37 |
Abrocitinib 100 mg N=84 |
p-value | |
|---|---|---|---|---|
| Total AEs, n (%) | 28 (23.1) | 14 (37.8) | 14 (16.7) | 0.01 |
| AEs leading to study discontinuation | 2 (1.6) | 0 (0) | 2 (2.4) | 0.34 |
| Abdominal pain | 7 (5.8) | 5 (13.5) | 2 (2.4) | 0.01 |
| Upper respiratory tract infection | 4 (3.3) | 1 (2.7) | 3 (3.6) | 0.79 |
| Headache | 4 (3.3) | 2 (5.4) | 2 (2.4) | 0.39 |
| Acne | 3 (2.5) | 2 (5.4) | 1 (1.2) | 0.17 |
| Nausea | 2 (1.7) | 1 (2.7) | 1 (1.2) | 0.54 |
| Weight gain | 2 (1.7) | 1 (2.7) | 1 (1.2) | 0.54 |
| Hypercholesterolaemia | 2 (1.7) | 0 (0) | 2 (2.4) | 0.33 |
| Herpes zoster | 1 (0.8) | 0 (0) | 1 (1.2) | 0.49 |
| Elevated liver enzymes | 1 (0.8) | 1 (2.7) | 0 (0) | 0.12 |
| Urinary tract infection | 1 (0.8) | 1 (2.7) | 0 (0) | 0.12 |
| Elevated CPK | 1 (0.8) | 0 (0) | 1 (1.2) | 0.49 |
|
aPercentage of patients with at least one follow up visit. AE:Adverse Events ; CPK:Creatine Phosphokinase. |
||||
DISCUSSION
This study demonstrates that abrocitinib is highly effective in adults with moderate-to-severe AD, providing clear or almost clear skin and significant relief from itch and sleep disturbance within 2 months, with further improvements up to 12 months. Effectiveness was observed even in patients previously treated with biologics and in difficult-to-treat areas. Approximately two-thirds of patients achieved MDA by 12 months. The safety profile was favourable, with no severe AEs, although abrocitinib 100 mg showed a better safety profile than 200 mg. These findings are consistent with existing clinical trial data and systematic reviews.
At week 16, clinical responses in our study were superior to those reported in Phase III trials, including JADE MONO-1, with higher rates of IGA 0/1 (74% vs 44%), EASI-75 (78% vs 63%) and EASI-90 (51% vs 39%), as well as greater pruritus improvement (ΔPP-NRS ≥4: 74% vs 57%) (10). However, JADE MONO-1 outcomes were assessed after 12 weeks of treatment with 200 mg abrocitinib.
In JADE EXTEND, the proportion of patients achieving EASI-90 and ≥4-point reduction in PP-NRS at week 48 was similar to week 12 (21). In contrast, our study showed a progressive increase in responders from week 8 to week 52, with improvements observed across all clinical outcomes, not limited to EASI-90 and PP-NRS≥4, consistent with JADE EXTEND results (21). Outcome rates at week 52 in our cohort were higher than those reported in JADE EXTEND at week 48 with abrocitinib 200 mg, including IGA 0/1 (88.9% vs 55.1%), EASI-75 (96.3% vs 85.5%), EASI-90, (79.6% vs 61.1%), EASI-100 (59.3% vs 24.1%), PP-NRS ≥4 (86.8% vs 69.4%) and PP-NRS 0/1 (73.5% vs 40.5%). Similarly to JADE EXTEND, in our study both 100 mg and 200 mg doses demonstrated clinically meaningful long-term efficacy through week 52 in the majority of patients, suggesting that dose may not substantially affect response rates. However, comparisons across these studies should be interpreted cautiously due to differences in assessment time points and concomitant topical corticosteroid use, that was not consistently controlled in our cohort. (10, 21)
In the JADE-DARE post hoc analysis, high-level responses across multiple domains (EASI-90, PP-NRS 0/1 and DLQI 0/1) were achieved in 24 % of abrocitinib-treated patients versus 12 % of dupilumab-treated patients at week 26 (22). Using the treat-to-target framework (18), which defines MDA as the simultaneous achievement of EASI-90 and PP-NRS≤1, our study showed progressive increases in MDA rates: 18 % at week 8, 42 % at week 16, 50 % at week 32 and 66 % at week 52, exceeding outcomes reported in the JADE-DARE (22).
The safety and tolerability profile of abrocitinib in our study aligned with previous JADE COMPARE findings, with most AEs being mild and manageable (23). The frequency of treatment-related AEs in our cohort was relatively low (23 % over 52 weeks), most commonly gastrointestinal symptoms (5.8%), upper respiratory tract infections (3.3%) and headache (3.3%). The 100 mg dose showed superior tolerability compared to 200 mg, particularly for abdominal pain (2.4% vs 13.5%; p=0.01), supporting a dose-dependent safety profile described in prior trials (23). Rates of upper respiratory tract infections and headache were similar to JADE COMPARE (23), whereas acne (2.5%) and other AEs occurred less frequently, with slightly higher rates in the 200 mg group (5.4%) compared with 100 mg (1.2%). Study discontinuation due to AEs was 1.6%, comparable to JADE COMPARE (0.8–1.4% with 100 mg and 200 mg, respectively). Our findings are in line with the long-term pooled safety analysis from the JADE program, which reported mild infections—such as upper respiratory tract, herpes zoster and herpes simplex infections—and gastrointestinal symptoms as the most frequently observed AEs. However, long-term pooled safety data from the JADE program (up to~~ years,>5,200 patient-years) reported additional risks such as venous thromboembolism, malignancies and lymphopenia, particularly in older patients or those with specific risk factors, which were not observed in our one-year follow-up (24).
Real-world evidence on abrocitinib remains limited (12, 25, 26, 27, 28, 29, 30, 31). Compared with a recent 52-week retrospective, single-centre Italian study on abrocitinib in patients with moderate-to-severe AD, our study showed slightly lower rates of EASI-75, -90, -100 and IGA 0/1, but higher rates of ΔPP-NRS≥ 4 and PP-NRS 0/1 (86.8% and 73.5% vs ≈80 % and ≈60%, respectively) at week 52. The marked antipruritic efficacy of abrocitinib is thought to result from its selective inhibition of JAK1, a pivotal mediator of cytokine networks implicated in AD, particularly those involving IL-31, IL-4, IL-13 and IL-22, all of which contribute to itch pathogenesis (32). AEs were similar across the two studies (23% vs 32%), with lower rates of acne (2.5% vs 8%), hypercholesterolaemia (1.7% vs 8%) and herpes zoster (0.8% vs 4%), whereas a clear dose-dependent increase in AEs with 200 mg was a key finding in our study.
The study’s limitations include its retrospective design, relatively small sample size, predominance of Caucasian patients, allowance of concomitant topical treatments and multicentre assessments by different clinicians, which may introduce bias, although reflective of real-world practice. Although smoking status could potentially influence treatment response, only 2.1% of patients in our cohort were active smokers; therefore, a statistically meaningful comparison between smokers and non-smokers could not be performed
In conclusion, abrocitinib is highly effective and safe in adults with moderate-to-severe AD, providing early and progressive clinical benefits sustained through one year. Longer-term studies are needed to confirm its role in the management of AD.
REFERENCES
- Wollenberg A, Kinberger M, Arents B, Aszodi N, Avila Valle G, Barbarot S, et al. European guideline (EuroGuiDerm) on atopic eczema: part I - systemic therapy. J Eur Acad Dermatol Venereol 2022; 36: 1409–1431. https://doi.org/10.1111/jdv.18345
- Liu H, Dong H, Chen W, Li X, Xiao Z, Li H. Global, regional, and national burden of atopic dermatitis: insights from the Global Burden of Disease Study 2021. Dermatitis 2025. https://doi.org/10.1177/17103568251365559
- Silverberg JI, Hong HCH, Calimlim BM, Lee WJ, Teixeira HD, Collins EB, et al. Comparative efficacy of targeted systemic therapies for moderate-to-severe atopic dermatitis without topical corticosteroids: an updated network meta-analysis. Dermatol Ther 2023; 13: 2247–2264. https://doi.org/10.1007/s13555-023-01000-3
- Wollenberg A, Kinberger M, Arents B, Aszodi N, Avila Valle G, Barbarot S, et al. European guideline (EuroGuiDerm) on atopic eczema - part II: non-systemic treatments and treatment recommendations for special AE patient populations. J Eur Acad Dermatol Venereol 2022; 36: 1904–1926. https://doi.org/10.1111/jdv.18429
- Simpson EL, Bieber T, Eckert L, Wu R, Ardeleanu M, Graham NMH, et al. Patient burden of moderate to severe atopic dermatitis (AD): insights from a phase 2b clinical trial of dupilumab in adults. J Am Acad Dermatol 2016; 74: 491–498. https://doi.org/10.1016/j.jaad.2015.10.043
- Drucker AM, Wang AR, Li WQ, Sevetson E, Block JK, Qureshi AA. The burden of atopic dermatitis: summary of a report for the National Eczema Association. J Invest Dermatol 2017; 137: 26–30. https://doi.org/10.1016/j.jid.2016.07.012
- Whiteley J, Emir B, Seitzman R, Makinson G. The burden of atopic dermatitis in US adults: results from the 2013 National Health and Wellness Survey. Curr Med Res Opin 2016; 32: 1645–1651. https://doi.org/10.1080/03007995.2016.1195733
- Chovatiya R, Paller AS. JAK inhibitors in the treatment of atopic dermatitis. J Allergy Clin Immunol 2021; 148: 927–940. https://doi.org/10.1016/j.jaci.2021.08.009
- Gargiulo L, Ibba L, Malagoli P, Burroni AG, Chiricozzi A, Dapavo P, et al. Management of patients affected by moderate-to-severe atopic dermatitis with JAK inhibitors in real-world clinical practice: an Italian Delphi consensus. Dermatol Ther 2024; 14: 919–932. https://doi.org/10.1007/s13555-024-01135-x
- Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet 2020; 396: 255–266. https://doi.org/10.1016/S0140-6736(20)30732-7
- Bieber T, Simpson EL, Silverberg JI, Thaçi D, Paul C, Pink AE, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med 2021; 384: 1101–1112. https://doi.org/10.1056/NEJMoa2019380
- Gargiulo L, Ibba L, Alfano A, Malagoli P, Amoruso F, Balato A, et al. Short-term effectiveness and safety of abrocitinib in adults with moderate-to-severe atopic dermatitis: results from a 16-week real-world multicenter retrospective study - il AD (Italian landscape atopic dermatitis). J Dermatolog Treat 2024; 35: 2411855. https://doi.org/10.1080/09546634.2024.2411855
- European Medicines Agency. Cibinqo (abrocitinib): summary of product characteristics. 2021. [cited 2025 August 28]. Available from: https: //www.ema.europa.eu/en/documents/overview/cibinqo-epar-medicine-overview_en.pdf
- World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human participants. JAMA 2025; 333: 71–74. https://doi.org/10.1001/jama.2024.21972
- STROBE statement. [cited 2023 March 1]. Available from: https://www.strobe-statement.org/checklists/cohort-studies
- Yosipovitch G, Reaney M, Mastey V, Eckert L, Abbé A, Nelson L, et al. Peak pruritus numerical rating scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol 2019; 181: 761–769. https://doi.org/10.1111/bjd.17744
- Schram ME, Spuls PI, Leeflang MMG, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy 2012; 67: 99–106. https://doi.org/10.1111/j.1398-9995.2011.02719.x
- Silverberg JI, Gooderham M, Katoh N, Aoki V, Pink AE, Binamer Y, et al. Combining treat‐to‐target principles and shared decision‐making: International expert consensus‐based recommendations with a novel concept for minimal disease activity criteria in atopic dermatitis. Acad Dermatol Venereol 2024; 38: 2139–2148. https://doi.org/10.1111/jdv.20229
- Drucker AM, Morra DE, Prieto-Merino D, Ellis AG, Yiu ZZN, Rochwerg B, et al. Systemic immunomodulatory treatments for atopic dermatitis: update of a living systematic review and network meta-analysis. JAMA Dermatol 2022; 158: 523–532. https://doi.org/10.1001/jamadermatol.2022.0455
- Ito T, Sugasawa S. Grouped generalized estimating equations for longitudinal data analysis. Biometrics 2023; 79: 1868–1879. https://doi.org/10.1111/biom.13718
- Shi VY, Bhutani T, Fonacier L, Deleuran M, Shumack S, Valdez H, et al. Phase 3 efficacy and safety of abrocitinib in adults with moderate-to-severe atopic dermatitis after switching from dupilumab (JADE EXTEND). J Am Acad Dermatol 2022; 87: 351–358. https://doi.org/10.1016/j.jaad.2022.04.009
- Silverberg JI, Simpson EL, Pink AE, Weidinger S, Chan G, Biswas P, et al. Switching from dupilumab to abrocitinib in patients with moderate-to-severe atopic dermatitis: a post hoc analysis of efficacy after treatment with dupilumab in JADE DARE. Dermatol Ther 2025; 15: 367–380. https://doi.org/10.1007/s13555-024-01320-y
- Simpson EL, Silverberg JI, Thyssen JP, Viguier M, Thaçi D, de Bruin-Weller M, et al. Efficacy and safety of abrocitinib in patients with severe and/or difficult-to-treat atopic dermatitis: a post hoc analysis of the randomized phase 3 JADE COMPARE trial. Am J Clin Dermatol 2023; 24: 609–621. https://doi.org/10.1007/s40257-023-00785-5
- Simpson EL, Silverberg JI, Nosbaum A, Winthrop K, Guttman-Yassky E, Hoffmeister KM, et al. Integrated safety update of abrocitinib in 3802 patients with moderate-to-severe atopic dermatitis: data from more than 5200 patient-years with up to 4 years of exposure. Am J Clin Dermatol 2024; 25: 639–654. https://doi.org/10.1007/s40257-024-00869-w
- Tong Z, Zhang Y, Zhou K, Zou Y, Wu Z, Chen J, et al. An observational study of abrocitinib in adults with moderate-to-severe atopic dermatitis after switching from dupilumab. J Am Acad Dermatol 2023; 89: 826–828. https://doi.org/10.1016/j.jaad.2023.05.077
- Olydam JI, Schlösser AR, Custurone P, Nijsten TEC, Hijnen D. Real-world effectiveness of abrocitinib treatment in patients with difficult-to-treat atopic dermatitis. J Eur Acad Dermatol Venereol 2023; 37: 2537–2542. https://doi.org/10.1111/jdv.19378
- Armario-Hita JC, Pereyra-Rodriguez JJ, González-Quesada A, Herranz P, Suarez R, Galan-Gutiérrez M, et al. Treatment of atopic dermatitis with abrocitinib in real practice in Spain: efficacy and safety results from a 24-week multicenter study. Int J Dermatol 2024; 63: e289–e295. https://doi.org/10.1111/ijd.17344
- Keow S, Abu-Hilal M. Effectiveness of abrocitinib for the treatment of moderate-to-severe atopic dermatitis in patients switched from dupilumab and/or tralokinumab: a real-world retrospective study. J Am Acad Dermatol 2024; 91: 734–735. https://doi.org/10.1016/j.jaad.2024.05.081
- Gargiulo L, Ibba L, Piscazzi F, Alfano A, Cascio Ingurgio R, Valenti M, et al. Effectiveness and safety of upadacitinib for moderate-to-severe atopic dermatitis in a real-world setting: a 52-week retrospective study. J Eur Acad Dermatol Venereol 2024; 38: e152–e154. https://doi.org/10.1111/jdv.19507
- Hu Q, Gao Y, Xu K, Yao X. Drug survival of abrocitinib compared to dupilumab in adult patients with atopic dermatitis. J Eur Acad Dermatol Venereol 2025; 39: e269–e270. https://doi.org/10.1111/jdv.20265
- Ibba L, Falcidia C, Di Giulio S, Bianco M, Valenti M, Facheris P, et al. Real-world effectiveness and safety of upadacitinib and abrocitinib in moderate-to-severe atopic dermatitis: a 52-week retrospective study. J Clin Med 2025; 14: 2953. https://doi.org/10.3390/jcm14092953
- Kim BS, Silverberg JI, Ständer S, Yosipovitch G, Simpson EL, DiBonaventura M, et al. Rapid improvement of itch associated with atopic dermatitis with abrocitinib is partially independent of overall disease improvement: results from pooled phase 2b and 3 monotherapy studies. Dermatitis 2021; 32: S39–S44. https://doi.org/10.1097/DER.0000000000000770