QUIZ SECTION
Swelling of the Left Upper Limb with Red Papules, Necrosis and Crusting: A Quiz
Jia-Wei LIU1†, Jie ZHANG1† and Hong-Zhong JIN1*
1Department of Dermatology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, National Clinical Research Center for Dermatologic and Immunologic, Beijing, China. *Email: jinhongzhong@263.net
†These authors contributed equally to this work.
Citation: Acta Derm Venereol 2026; 106: adv-2025-0120. DOI: https://doi.org/10.2340/actadv.v106.adv-2025-0120.
Copyright: © 2026 The Author(s). Published by MJS Publishing, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/).
Submitted: Accepted after revision:
Published: May 25, 2026.
Competing interests and funding:
A man in his 40s presented to our clinic with progressive swelling and sclerosis of his left upper limb. The lesion first appeared as swelling on the dorsum and palm of his hand 8 months ago. The swelling gradually extended to the upper arm, with erythematous papules, pustules, erosion and crusting in a band-like pattern (Figs 1 and 2). The patient denied tenderness or pain on movement. Laboratory tests showed low white blood cell counts of 2.09×10⁹/L (normal range 4–10×10⁹/L) and neutrophil counts of 1.24×10⁹/L (normal range 2–7×10⁹/L), while liver function, kidney function, antinuclear antibody, antineutrophil cytoplasmic antibody and inflammatory markers were normal. Tumour marker test revealed an elevated cancer antigen 125 (CA125) level of 142 U/mL (normal range <35 U/mL). Magnetic resonance imaging revealed osteomyelitis of the second and third metacarpals, thickened fascia and subcutaneous oedema. Positron emission tomography/computed tomography showed mild fluorodeoxyglucose uptake in the affected areas. Tissue cultures and next-generation sequencing identified Pseudomonas aeruginosa, but empirical antimicrobials, including moxifloxacin, rifampin, clarithromycin, minocycline, meropenem, ciprofloxacin, fluconazole and voriconazole, were ineffective. Biopsy results from the upper arm are shown in Fig. 3.
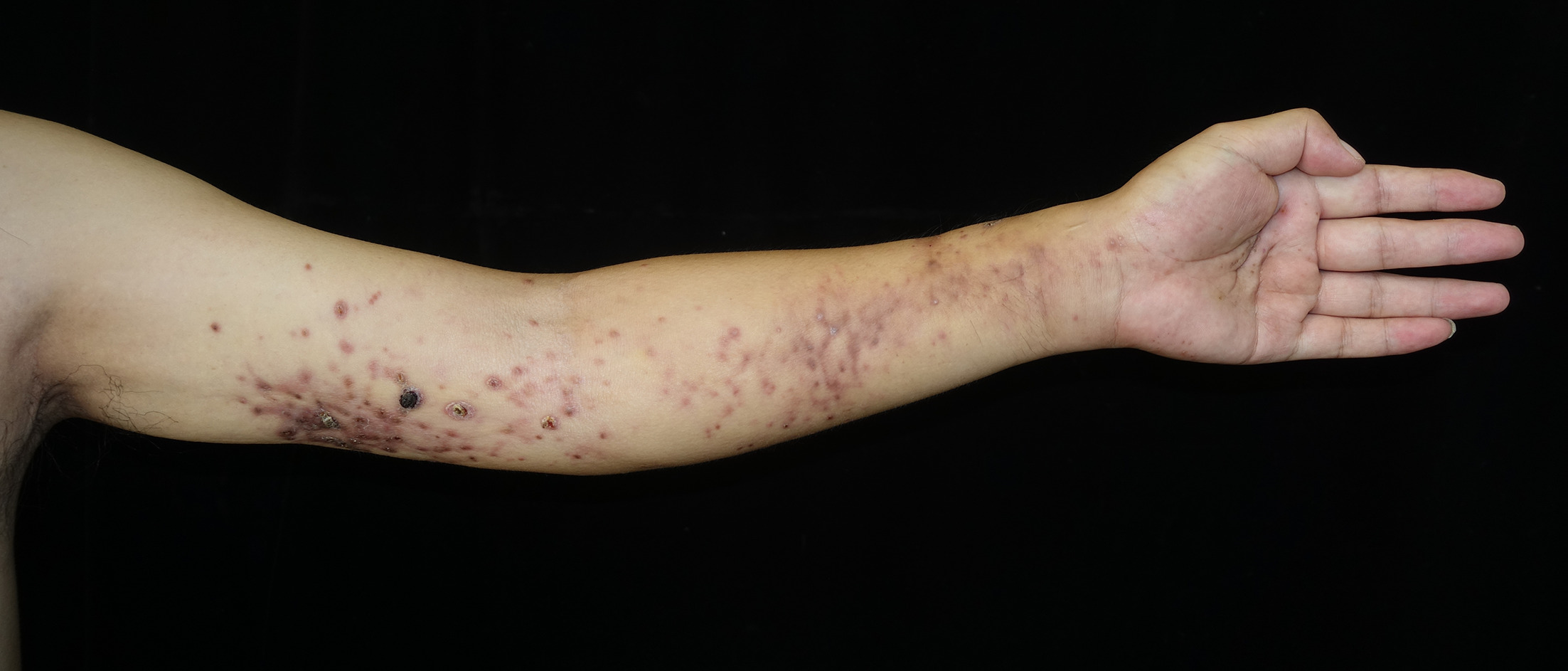
Fig. 1. Skin lesion on the left upper limb: Swelling and skin sclerosis of the left upper limb with red papules, crusting and necrosis in a band-like distribution.
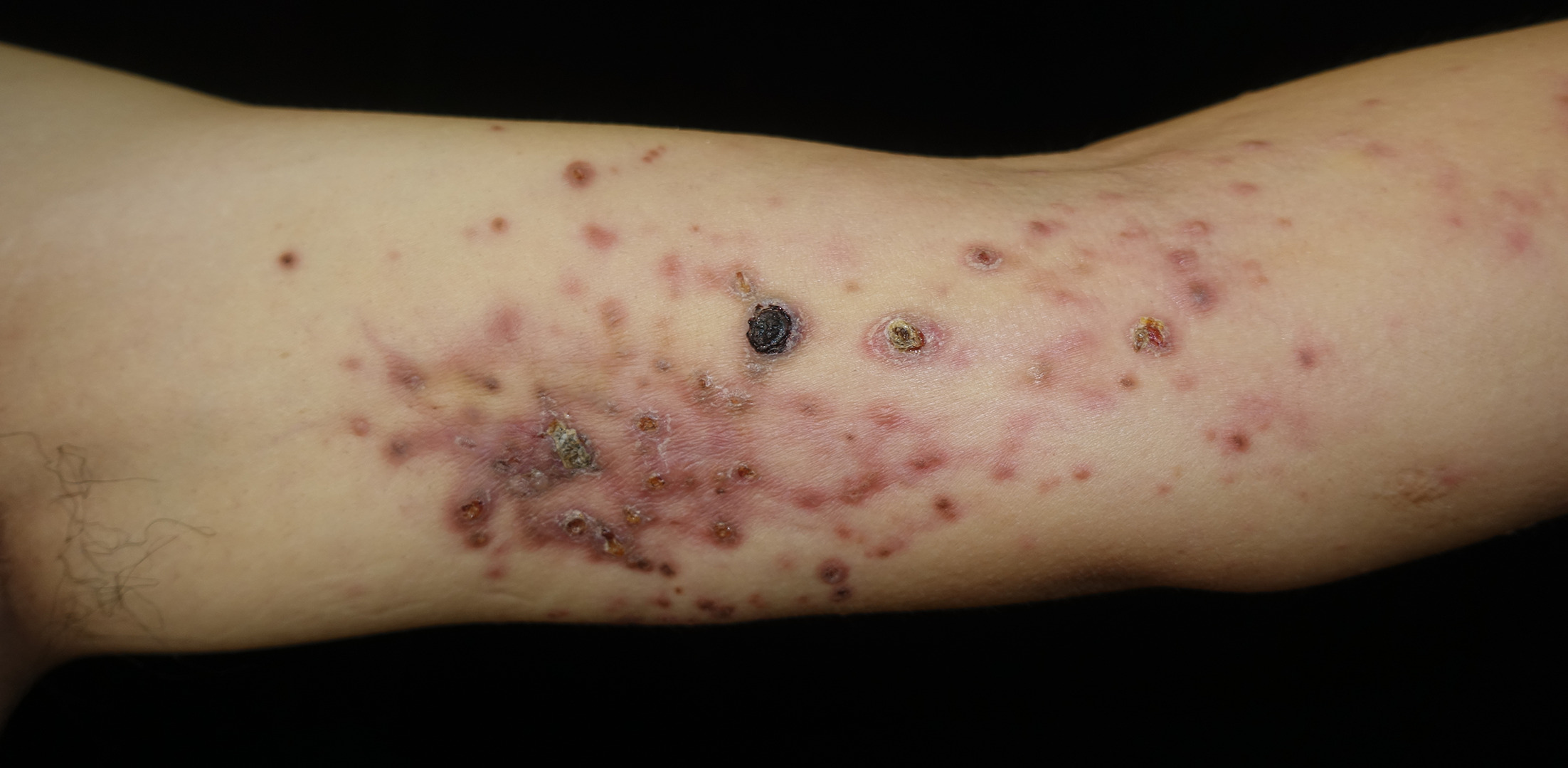
Fig. 2. Close examination of the skin lesion on the upper arm: Multiple erythematous papules partially coalesce, punctate necrosis and crusting, and some necrotic lesions can heal spontaneously, leaving atrophic scars.
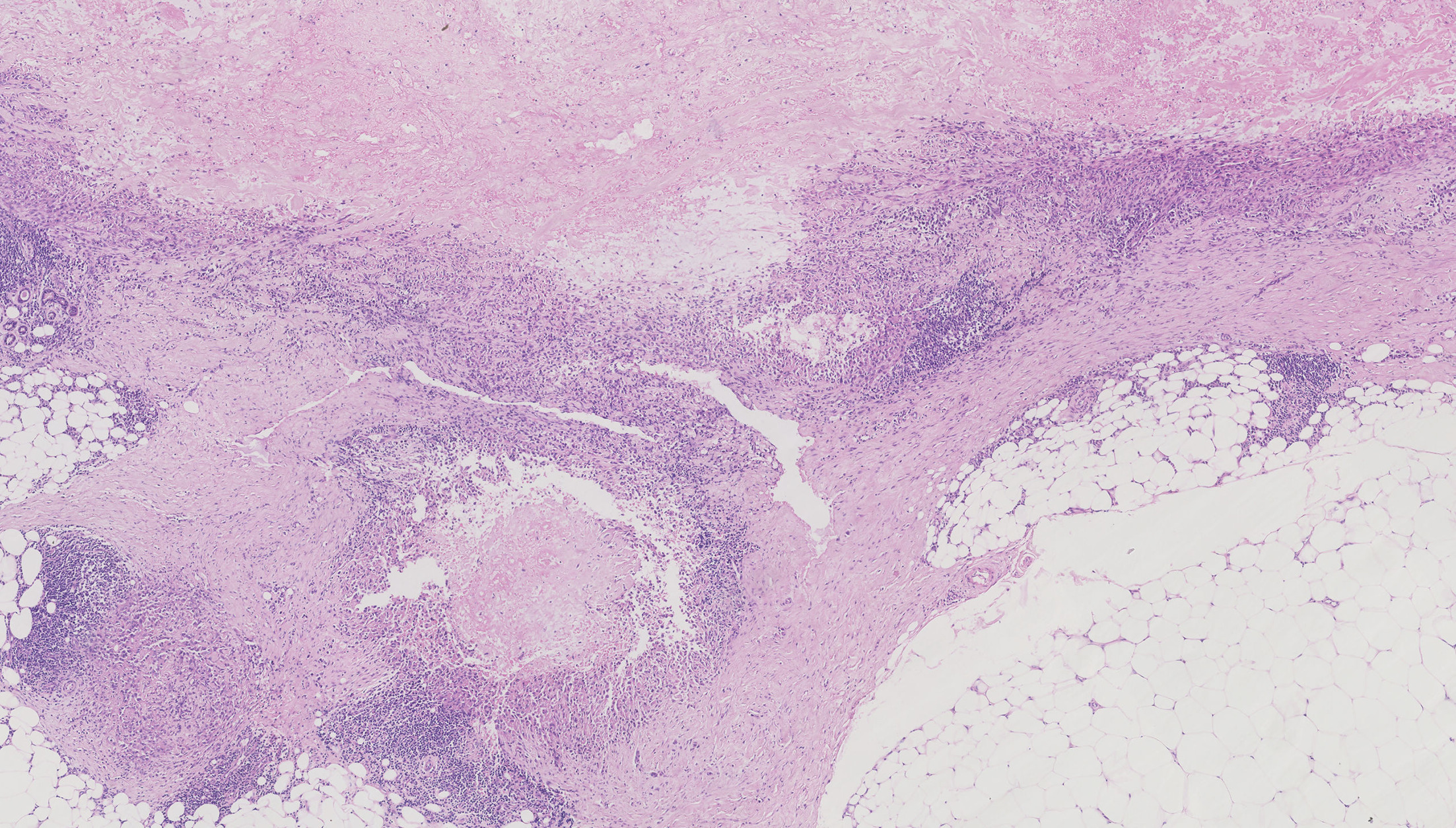
Fig. 3. Histological finding of the skin lesion: Skin biopsy revealed a granulomatous pattern, with epithelioid cells with abundant eosinophilic cytoplasm arranged in a fascicular pattern, encircling a central necrotic area.
What is your diagnosis?
1: Nontuberculous mycobacteria (NTM) skin infection
2: Sporotrichosis
3: Epithelioid sarcoma
4: Epithelioid malignant peripheral nerve sheath tumour
See next page for answer.
ANSWERS TO QUIZ
Swelling of the Left Upper Limb with Red Papules, Necrosis and Crusting: A Commentary
Diagnosis: Epithelioid Sarcoma
Histopathology revealed a granulomatous pattern with central necrosis, spindle cells and epithelioid cells (Fig. 3). Immunohistochemical staining was positive for vimentin, cytokeratin (CK), epithelial membrane antigen (EMA) and CD34, with a Ki-67 index of 10%. The diagnosis was distal-type epithelioid sarcoma (ES), and the patient was referred to oncology for further treatment.
ES is a rare and aggressive soft tissue malignancy, accounting for less than 1% of all soft tissue sarcomas. ES predominantly affects young adults with a peak incidence around 35 years of age and demonstrates a male predominance with a male-to-female ratio of 2 : 1 (1).
ES is classified into two main subtypes: distal (classic) type and proximal-type ES. The distal type typically affects the extremities, particularly the hands and forearms of adolescents and young adults (2). It manifests as slow-growing, painless, firm nodules that may evolve into non-healing ulcers. These lesions often develop in the dermis or subcutaneous tissue, but can also occur deeply in tenosynovial tissues, with the potential to spread along nerves and fascial planes (3). The distal variant follows a characteristic pattern of repeated local recurrences, with successive lesions often arising more proximally. This clinical behaviour distinguishes it from the more aggressive proximal-type ES, which affects deeper tissues of proximal extremities and the trunk in slightly older patients (4).
Histologically, classic ES demonstrates a characteristic appearance of nodular aggregates of fairly uniform, plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central zonal necrosis, creating a pseudogranulomatous appearance. Cellular atypia is generally mild. Spindled cells may be present focally, particularly at the periphery of the lesions (5).
Immunohistochemically, ES displays features of both epithelial and mesenchymal differentiation. Nearly all cases express CK and EMA, while approximately half express CD34 and CA125. The most significant diagnostic immunohistochemical marker is the loss of nuclear integrase interactor 1 expression in approximately 90% of cases (6). Serum CA125 is also elevated and could be utilized as a diagnostic and management marker for the disease.
The differential diagnosis includes both benign and malignant conditions. Non-neoplastic conditions such as necrotizing granulomas, granuloma annulare and rheumatoid nodules must be excluded. Malignant entities in the differential include other INI1-deficient tumours (malignant rhabdoid tumour, epithelioid malignant peripheral nerve sheath tumour), epithelioid angiosarcoma, pseudomyogenic haemangioendothelioma, myoepithelial tumours, poorly differentiated synovial sarcoma and metastatic carcinoma (7). The integration of clinical, morphological and immunohistochemical features is crucial for accurate diagnosis.
The mainstay of treatment for localized ES remains wide surgical excision with clear margins (8). Adjuvant or neoadjuvant radiotherapy may reduce local recurrence risk, particularly for high-risk tumours (7). The sentinel lymph node biopsy should be considered given the relatively high rate of lymph node metastasis (up to 30%) (9).
For advanced disease, systemic therapy options are limited. Anthracycline-based regimens show response rates of approximately 22%, while gemcitabine-based regimens reported response rates of 27–58% in different series (9). Tazemetostat, an enhancer of zeste homology 2 inhibitor, was approved for metastatic or locally advanced ES not eligible for complete resection, with a median progression-free survival of 11.1–13.8 months (10).
Other potential therapeutic targets under investigation include the PI3K (phosphatidylinositol 3-kinase)/AKT (protein kinase B)/mTOR (mechanistic target of rapamycin) pathway, EGFR (epidermal growth factor receptor), c-MET (MET proto-oncogene, receptor tyrosine kinase) and immune checkpoints. Despite these advances, the prognosis for metastatic ES remains poor, with a median overall survival of approximately 18 months (10), highlighting the importance of early diagnosis as well as novel targeted approaches in improving outcomes in ES patients.
REFERENCES
- Czarnecka AM, Sobczuk P, Kostrzanowski M, Spalek M, Chojnacka M, Szumera-Cieckiewicz A, et al. Epithelioid sarcoma – from genetics to clinical practice. Cancers 2020; 12: 2112. https://doi.org/10.3390/cancers12082112
- Chase DR, Enzinger FM. Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am J Surg Pathol 1985; 9: 241–263.
- Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol 2000; 7: 218–225. https://doi.org/10.1007/BF02523657
- Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol 1997; 21: 130–146. https://doi.org/10.1097/00000478-199702000-00002
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol 2006; 13: 114–121. https://doi.org/10.1097/00125480-200605000-00002
- Hornick JL, Dal Cin P, Fletcher CDM. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 2009; 33: 542–550. https://doi.org/10.1097/PAS.0b013e3181882c54
- Thway K, Jones RL, Noujaim J, Fisher C. Epithelioid sarcoma: diagnostic features and genetics. Adv Anat Pathol 2016; 23: 41–49. https://doi.org/10.1097/PAP.0000000000000102
- Wolf PS, Flum DR, Tanas MR, Rubin BP, Mann GN. Epithelioid sarcoma: the University of Washington experience. Am J Surg 2008; 196: 407–412. https://doi.org/10.1016/j.amjsurg.2007.07.029
- Alves A, Constantinidou A, Thway K, Fisher C, Huang P, Jones RL. The evolving management of epithelioid sarcoma. Eur J Cancer Care 2021; 30: e13489. https://doi.org/10.1111/ecc.13489
- Gounder MM, Merriam P, Ratan R, Patel SR, Chugh R, Villalobos VM, et al. Real-world outcomes of patients with locally advanced or metastatic epithelioid sarcoma. Cancer 2021; 127: 1311–1317. https://doi.org/10.1002/cncr.33365