ORIGINAL REPORT
Characteristics of Café-au-lait Macules and their Association with the Neurofibromatosis type I Genotype in a Cohort of Greek Children
Lamprini NASI1, Alexios ALEXOPOULOS1, Eleftheria KOKKINOU2, Kleoniki ROKA3, Maria TZETIS4, Maria TSIPI4, Talia KAKOUROU1, Christina KANAKA-GANTENBEIN5, George CHROUSOS5, Antonis KATTAMIS3, Roser PONS2 and the Agia Sophia Neurocutaneous Group*
1Unit of Pediatric Dermatology, 2Division of Pediatric Neurology, 3Division of Pediatric Hematology-Oncology, First Department of Pediatrics, Medical School, National and Kapodistrian University of Athens, Agia Sophia Children’s Hospital, Athens, Greece, 4Laboratory of Medical Genetics, Medical School and 5First Department of Pediatrics, Medical School, National and Kapodistrian University of Athens, Agia Sophia Children’s Hospital, Athens, Greece
Cafe-au-lait macules are the most distinctive clinical finding in neurofibromatosis type I. The aim of this prospective study of Greek children diagnosed with neurofibromatosis type I was to describe the dermatological phenotype and to analyse the characteristics of cafe-au-lait macules and their association with genotype. Pigment intensity and melatonin content of cafe-au-lait macules were measured with a narrowband spectrophotometer. A total of 63 children aged 6 months to 16 years old were studied. Mean melanin content varied, both among patients, and within each patient (p < 0.001). Females had a higher number of cafe-au-lait macules than did males (p = 0.025), and the melanin content of cafe-au-lait macules was lower in females than males (p < 0.001). Patients with protein-truncating variants in the neurofibromatosis type I gene had higher melanin content of cafe-au-lait macules than other types of genetic variants t (55) = 2.196, p = 0.032. Plexiform neurofibromas were also detected in the majority of patients with protein- truncating variants, while juvenile xanthogranulomas were detected equally in patients with protein-truncating and non-protein-truncating variants. In conclusion, cafe-au-lait macules with high melatonin content are associated with patients carrying non-protein-truncating variants. Therefore, measurement of cafe-au-lait macule pigment intensity might provide useful information for initial assessment of patients with neurofibromatosis type I and the severity of their future phenotype.
Key words: neurofibromatosis; café-au-lait macules; pigment intensity; melanin; genotype.
SIGNIFICANCE
Cafe-au-lait macules are the most frequent clinical manifestation in neurofibromatosis type 1 and represent a major criterion for diagnosis of neurofibromatosis type 1. This study established the variability of melanin content among patients with neurofibromatosis type 1 and within each individual. The study concluded that patients with neurofibromatosis type 1 with protein-truncating variants that are likely to lead to severe future phenotype have significantly higher melatonin content of cafe-au-lait macules than patients with non-protein-truncating variants. The study also found that the majority of patients with plexiform neurofibromas carried protein-truncating variants, while juvenile xanthogranulomas were detected equally in patients with protein-truncating and non-protein-truncating variants.
Citation: Acta Derm Venereol 2023; 103: adv5758. DOI: https://doi.org/10.2340/actadv.v103.5758.
Copyright: © Published by Medical Journals Sweden, on behalf of the Society for Publication of Acta Dermato-Venereologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/)
Accepted: May 8, 2023; Published: Jun 5, 2023
Corr: Nasi Lamprini, First Department of Pediatrics, Medical School, National and Kapodistrian University of Athens, “Agia Sophia” Children’s Hospital, Thivon & Papadiamantopoulou Street, GR-11527 Athens, Greece. E-mail: nasi_lina@yahoo.gr
Competing interests and funding: The authors have no conflicts of interest to declare.
INTRODUCTION
Neurofibromatosis type 1 (NF1) is a multisystemic autosomal dominant genetic disorder, with a birth incidence of 1:2000 and a prevalence of 1/4000 (1, 2). The most prominent clinical manifestations of NF1 are café au lait macules (CALMs) and neurofibromas (subcutaneous, cutaneous, and plexiform), which are present in more than 95% of adult patients (3). Two out of 7 major diagnostic criteria are required to determine the diagnosis, with the presence of 6 or more CALMs larger than 5 mm before puberty and larger than 15 mm in adults being the most widely recognized among them (4). Other cutaneous manifestations of NF1 include freckling, juvenile xanthogranuloma (JXG), naevus anaemicus (NA) and glomus tumours (5).
CALMs are often the first manifestation of NF1. They are described as uniform, light- to dark-brown in colour, oval in shape, and smooth in texture macules (“California coast” type) (6), and their distribution is random, excluding the scalp, palms and soles (7, 8). CALMs may be present at birth; they gradually increase in size and number during the first years of life; however, most CALMs usually emerge by the age of 2–4 years. The incidence of CALMs in patients with NF1 is high; therefore, their detection and assessment are important for the early diagnosis of NF1 (9–11).
Most patients with NF1 carry mutations within the NF1 gene. The genomic location of the NF1 gene is 17q11.2 and it consists of 58 exons. This large gene holds one of the highest genetic variant rates in the entire human genome (12). Several types of genetic variants associated with NF1 phenotype have been described (13). De novo genetic variants occur in more than 50% of patients (3).
The NF1 gene encodes the protein “neurofibromin”, which has tumour-suppressive activity in many cells, notably neurones, ganglion cells, Schwann cells and early melanocytes (14). NF1 gene dysfunction is associated with a variable phenotype that includes tumour predisposition and it is hypothesized that the rate of growth of specific tumours is related to the nature of a “second hit” somatic genetic variant (6).
The pathogenesis of CALMs in NF1, and their correlation with the NF1 genotype has been insufficiently studied (15, 16). In 2008, De Schepper et al. (17) identified somatic genetic variants in the NF1 gene in melanocyte cultures from CALMs of patients with NF1, while only germline genetic variants were found in non-CALM skin melanocytes. Boyd et al. (18) hypothesized that the differences in pigmentation of CALMs may be related to the combination of a germline mutation and a second somatic genetic variant in specific melanocytes. This was supported by the finding of significant intra-individual variability of pigment intensity in the CALMs of patients with NF1.
The aim of this study was to analyse the dermatological findings of patients with genetically confirmed NF1, with an emphasis on CALMs pigmentation and its association with the NF1 genotype.
MATERIALS AND METHODS
This is a prospective study conducted between April 2018 and March 2022 at the Neurocutaneous Clinic of the First Department of Pediatrics of the National and Kapodistrian University of Athens Medical School at “Agia Sophia” Children’s Hospital of Athens in Greece, which was established in 2016 (19). The current study was designed and conducted in accordance with the principles of the Declaration of Helsinki and was approved by the ethics committee of First Department of Pediatrics of the National and Kapodistrian University of Athens Medical School at “Agia Sophia” Children’s Hospital of Athens in Greece (study protocol 23-02-18/4559). Informed consent was obtained from legal guardians of the children.
Inclusion criteria were: (i) patients with NF1 between 6 months to 16 years old; (ii) genetic information regarding the NF1 diagnosis. Genotyping of the cohort of patients was conducted by the Department of Medical Genetics, Medical School, National and Kapodistrian University of Athens, Greece. Some of the molecular findings have been published previously (19).
Chart review included sex, age, race, demographic and social data, family history, clinical data, and molecular findings.
CALM pigmentation measurements using a narrowband spectrophotometer were performed during regular outpatient visits of patients with NF1 at Neurocutaneous Clinic of the First Department of Pediatrics of the National and Kapodistrian University of Athens Medical School at “Agia Sophia” Children’s Hospital of Athens, Greece. The narrowband spectrophotometer calculates the relative absorption of melanin based on the absorption of visible light and, at the same time, provides satisfactory measurements of erythema and pigmentation rates. All colometric data were obtained using a narrowband spectrophotometer DSM III (Cortex Technology, Hadsund, Denmark). DSM III is a handheld instrument used for measurement of skin colour, including melanin. The melanin values from the DSM III are index values and are used for the estimation of level of pigment in the skin. Quantification of melanin content is a relative number comparing the data obtained with the DSM III of normal skin with the data of CALM. The DSM III records colour using 3 values developed by the Commission Internationale d’ Eclairage (CIE): L*, light intensity; a*, amount of green or red; and b*, amount of blue or yellow (18). Measurements were performed by 1 author (LN). Three successive measurements were performed for each measured CALM. Accordingly, 3 more measurements of the non-CALM skin were made in a 120° angle, to ensure consistency of measurements. The pigmentation of 6 CALMs per patient was measured. The selection of CALMs measured was by a random criterion.
All patients included in the study presented with type III photo type, according to Fitzpatrick scale (20). All measurements took place during the winter-time, on the covered areas of the skin, in order to ensure homogeneity of patients. A digital camera was used to determine the number and distribution of CALMs. The whole body of participants was photographed with a digital camera (Sony, DSC-H50, San Diego, USA). CALMs larger than 3 cm were mapped on a standard body chart, in order to report CALM body distribution (Fig. 1). All CALMs were classified into 4 categories based on their distribution: (i) head and neck, (ii) thorax, shoulder surface, (iii) upper and lower extremity, and (iv) inguinal area.
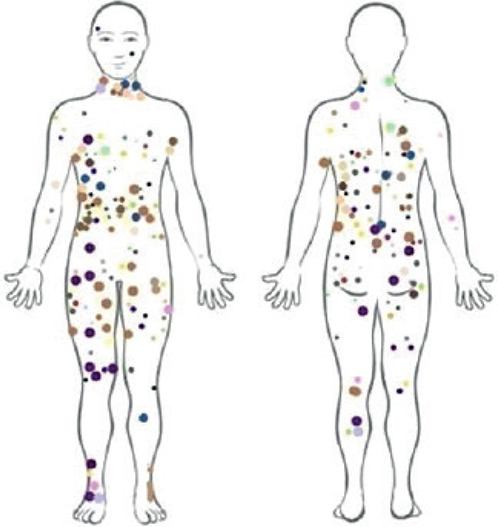
Fig. 1. Distribution of café-au-lait macules (larger than 3 cm) in the current series of 63 patients (each colour represents a unique individual).
Statistical analysis
For descriptive statistical analysis, continuous variables are presented as mean value, standard deviation, median and interquartile range (IQR: interquartile range), while discrete variables are presented as frequency (N) and relative frequency (N %). To control the condition of normality, the graphs “Normal Q-Q plot”, “Detrended Normal Q-Q plot” and “Box plot” were used. Levene’s test was applied to check the equality of population variations. Mood’s median test was used to check the median’s equality of more than 2 independent populations. For the comparative study of the mean values of a continuous variable, between 2 independent groups, the Mann–Whitney test was used. Spearman’s correlation coefficient was used to investigate the existence of a relationship between 2 quantitative variables. To study the relationship between 2 categorical variables, the χ2 test of independence was applied. SPSS software (25th edition; IBM, Chicago, IL, USA) was used to statistically evaluate the data. All tests were 2-tailed. Significance was set at p < 0.05.
RESULTS
The study included 63 children (33 boys and 30 girls) with genetically confirmed NF1. Mean (standard deviation; SD) and median age were 7.5 (4.6) and 8 (0.5–16) years, respectively. Their main clinical manifestations are shown in Table I.
All children with NF1 showed more than 6 CALMs, with a mean (SD) of 18.1 (8.3) and a median of 17 (Table II). Of these CALMs, 53.6% were located on the thorax, 9.1% on the upper extremities, 18.6% on the lower extremities, 13.4% on the inguinal area, and 5.4% on the head/neck (Table III). Axillary freckling was seen in 63.5% of patients, and inguinal freckling in 60.3% (Table I). Spearman correlation analysis showed no correlation between the total number of CALMs and age [rs(63) = –0.055, p = 0.666].
Mean CALMs diameter was found to be the largest in the chest area compared with other body sites. Kruskal–Wallis analysis of CALMs size and anatomical location showed significant differences χ2 (4) = 3.912, p < 0.001. Mean CALMs diameter was found to be the largest in the chest area compared with other body sites. Kruskal–Wallis analysis of CALMs size and anatomical location showed significant differences χ2 (4) = 41.266, p < 0.001.
Melanin content of CALMS is shown in Table II. Mood’s median test revealed that the mean value of melanin content among patients differed significantly [χ2 (5\9) = 208, p < 0.001], while Levene’s test showed that there was no homogeneity in CALM melanin content within each patient [F (59.42) = 1.972, p = 0.012]. Spearman correlation analysis showed a negative correlation between CALMs size and melanin content [rs (378) = –0.403, p < 0.001] (Fig. 2).
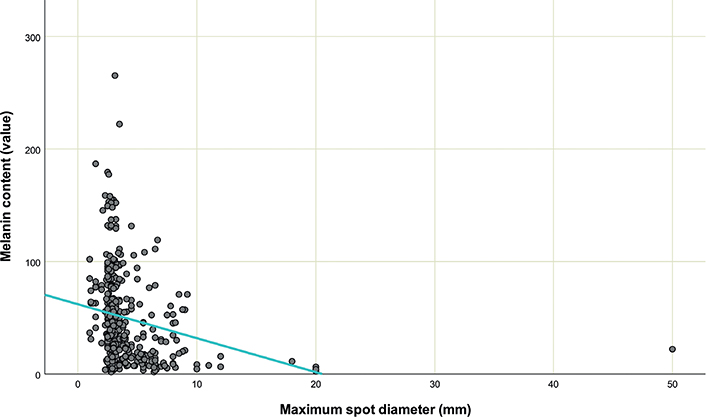
Fig. 2. Cafe-au-lait macules size and melanin content.
Melanin content in boys and girls is shown in Table II. Mean value of melanin content differed significantly between girls (N = 144, mean = 40.9, SD = 40.7) and boys (N = 234, mean = 54.8, SD = 41.3), t(376) = 3.194, p = 0.002. Mann–Whitney test showed significant difference in the total number of spots by sex [U (N boys = 33, N girls = 30) = 658, z = 2.246, p = 0.025], with girls having a higher total number of CALMs than boys. Levene’s test showed heterogeneity of CALMs melatonin content in girls [F (23, 120) = 14.284, p < 0.001] and in boys [F (38, 195) = 8.653, p < 0.001] (Fig. 3).
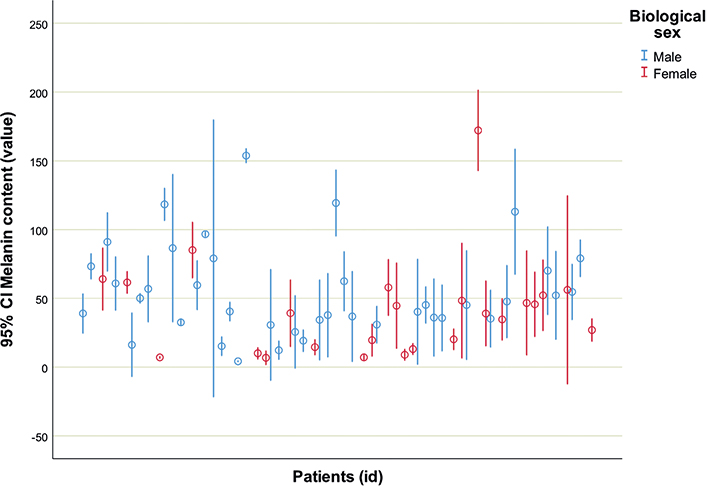
Fig. 3. Range of melanin content in patients with neurofibromatosis type 1 (N = 63). 95% CI: 95% confidence interval.
The molecular findings of this cohort of patients are shown in Table SI. Based on the severity of the genetic variants, 2 groups were delineated: group A with protein-truncating variants (nonsense and splice site (frame-shift) variants), and group B with non-protein-truncating variants (missense, splice site (in-frame) variants). In group A the mean melanin content was 50.6 (SD = 39.2) while in group B it was 34.6 (SD = 18.3). Independent t-test show statistically significant differences in the mean values of melanin content in the CALMs of these 2 groups t (55) = 2.196, p = 0.032 (Fig. 4).
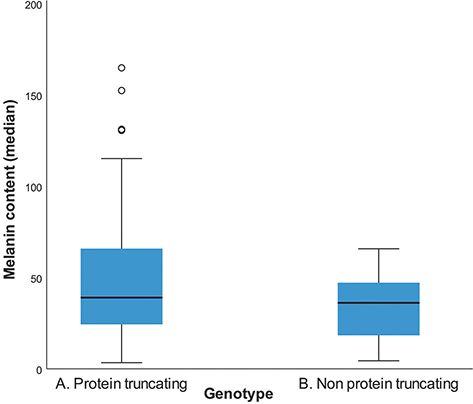
Fig. 4. Melanin content of cafe-au-lait macules per genotype group, group A (nonsense and frameshift variants), and group B (missense, splicing, in frame variants).
Cutaneous and subcutaneous neurofibromas were detected in 9 patients (15%): 30 in the thorax, 14 in the upper extremities, 3 in the lower extremities, and 2 in the inguinal area. Superficially located PNs were detected in 17 patients (27%). Seven PN were located in the head/neck area, 6 in the thorax, 2 in the upper extremities, and 2 in the lower extremities (Table III). Four patients had JXG (4 in the head, 1 in the face and 1 in the thorax), and 2 patients had NA (1 in the thorax and 1 in the upper limbs).
In the group of patients with JXG, the median (interquartile range; IQR) melanin content of CALMs was 16.8 (9.2–45.9), and in the group with external PNs it was 41.2 (30.0–59.5) . The Mann–Whitney U test did not show significant differences in the mean values of CALM melanin content in these 2 groups [U (NJXG = 3, NPNs = 17) = 36, z = 36, p = 0.266]. Fisher’s exact test did not reveal a statistically significant correlation between genotype group and the presence of JXG (p = 1.000). χ2 independence test did not reveal a statistically significant relationship between genotype group and the presence of PNs [×2 (1) = 0.838, p = 0.360].
DISCUSSION
To our knowledge, this is the first study of CALMs phenotype in the largest cohort of children who are all genetically confirmed with NF1 (Table SI).
CALMs are the most frequent clinical manifestations in NF1 and constitute a major diagnostic criterion of this disorder. In the current cohort of patients with NF1, the number of CALMs was notably higher than 6 (mean 16.2), which is set in the literature as the minimum number of CALMs for a diagnosis of NF1. This finding is consistent with earlier studies (21).
It has been hypothesized that CALMs in NF1 are the result of a germline loss of function mutation in the NF1 gene together with a somatic genetic variant of the NF1 gene in early skin melanocytes (21, 22). Earlier studies analysing melatonin content and searching for NF1 somatic alterations in melanocytes of CALMS in patients with NF1 supported this hypothesis, but both the number of patients and the number of CALMs studied were not sufficient to provide robust evidence (16–18).
The current study also demonstrated the presence of significant inter-individual variability of melanin content in CALMs of the patients with NF1, and that NF1 patients with protein-truncating variants have significantly higher CALM melatonin content than patients with non-protein-truncating variants. These findings indicate that protein-truncating variants might lead to more severe dysregulation of melanogenesis.
In concordance with our results Boyd et al. also showed significantly darker spots in individuals with germline genetic variants leading to haploinsufficiency, although only 7 out of the 24 patients in their study were genetically tested (18).
An interesting finding in the current cohort was the presence of more CALMs in females and higher melatonin content in males (Table II). These findings could be indicative of sex influences in the cutaneous manifestation of NF1. In fact, this is supported by studies showing that sex is a major prognostic factor underlying neuronal dysfunction in NF1, and suggests that sex should be taken into consideration when interpreting future preclinical and clinical studies (23).
Genotype-phenotype correlations have been reported in some patients with NF1 (15, 24). Koczkowska et al. (25) also detected a higher frequency of spinal bilateral neurofibromas in NF1 patients carrying missense genetic variants affecting codons 844 to 848. Kehrer-Sawatzk et al. reported a higher frequency of facial dysmorphisms, axillary and inguinal freckling and skeletal anomalies in NF1 patients carrying large deletions of the NF1 gene (2, 26). Kang et al. (27) demonstrated that patients with truncating and splicing genetic variants and large deletions often show a more severe NF1 clinical phenotype than patients with missense genetic variants.
In the current cohort of genetically confirmed NF1, the current study analysed potential genotype correlations with the presence of juvenille xanthogranuloma (JXG) and external plexiform neurofibroma (EPN). JXG is the most common form of non-Langerhans-cell histiocytosis and is characterized by benign yellowish to brownish papules (5). In NF1, JXG are reported up to 35% of cases and they may appear as early as CALMs (5). In the current cohort of patients with NF1, only 4 (6.7%) had evidence of JXG of them carried protein-truncating and the other 2 carried non protein-truncating variants.
PN are seen in 20–30% of patients with NF1. They are congenital Schwann cell-derived tumours that arise from somatic genetic variants of the wildtype NF1 allele (28). PN can be found either internally, where they cannot be found by physical examination, or they can be more superficially located. Superficially located PNs often manifest with enlargement of soft tissue with a “wrinkled” texture or a patch of hyperpigmentation with or without hypertrichosis (1). In the current cohort of patients with NF1, superficially located PNs were present in 16 patients (25.4%) and the majority carried protein-truncating variants. The current study did not include internal PNs in the analysis because in the current cohort not all patients had undergone comprehensive imaging in search for internal PNs. Since the majority of the current cohort were prepubertal, subcutaneous or peripheral nodular neurofibromas were not present. Consequently, the association of genotype with the presence of subcutaneous or peripheral nodular neurofibromas was also not performed, since this NF1 manifestation usually develops during adolescence.
Other cutaneous manifestations associated with NF1 include freckles and NA. Axillary and/or inguinal freckling constitutes a cardinal diagnostic feature of NF1 (1). They are small clustered pigmented macules (1–3 mm) that appear later than CALMs and they are found in more than 90% of patients with NF1 by the age of 7 years. We recorded axillary and/or inguinal freckling in 63.5% of patients in the current cohort, which was probably due to the younger median age of the current study population. In the current cohort, NA was detected in only 2 patients, although it has been reported in up to 50% of patients with NF1 (5, 29). The low frequency of NA in the current cohort of patients is unexplained and may be related to ethnic differences. Assessment of a larger cohort of patients will be necessary to better understand the basis of this observation.
Strengths of the current study were the number of patients with NF1 included and the fact that they were all genotyped. Furthermore, the current study used a tool, the DSM III that is effective and reliable for evaluating CALM pigmentation (30). Previous studies using this model have demonstrated the reliability and efficacy of measuring skin pigmentation. This study had some limitations: the single investigator who performed the test was not blinded to NF1 status; therefore, the potential of bias existed. In addition, the study measured only 6 CALMs for each patient, due to patients’ compliance issues.
In conclusion, this study demonstrates that, in patients with NF1, protein-truncating genetic variants in the NF1 gene are associated with higher CALMs melatonin content, while other cutaneous manifestation, such as JXG and PN, did not show any association with the type of genetic variant. If the current results are verified in future studies, measurement of pigment intensity in CALMs could be implemented in resource-limited settings, as a rapid, easy, risk-free test for the initial assessment of patients with NF1 and the severity of their future phenotype.
REFERENCES
- Bergqvist C, Servy A, Valeyrie-Allanore L, Ferkal S, Combemale P, Wolkenstein P, et al. NF France Network. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020; 15: 37.
- Kallionpää RA, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med 2018; 20: 1082–1086.
- Yao R, Wang L, Yu Y, Wang J, Shen Y. Diagnostic value of multiple café-au-lait macules for neurofibromatosis 1 in Chinese children. J Dermatol 2016; 43: 537–542.
- DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000; 105: 608–614.
- Ozarslan B, Russo T, Argenziano G, Santoro C, Piccolo V. Cutaneous findings in neurofibromatosis type 1. Cancers (Basel) 2021; 13: 463.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol 2009; 61: 1–16.
- Landau M, Krafchik BR. The diagnostic value of café-au-lait macules. J Am Acad Dermatol 1999; 40: 877–892.
- Friedman JM. Neurofibromatosis 1: clinical manifestations and diagnostic criteria. J Child Neurol 2002; 17: 548–651.
- Nunley KS, Gao F, Albers AC, Bayliss SJ, Gutmann DH. Predictive value of café au lait macules at initial consultation in the diagnosis of neurofibromatosis type 1. Arch Dermatol 2009; 145: 883–887.
- Ortonne JP, Brocard E, Floret D, Perrot H, Thivolet J. Valeur diagnostique des taches café-au-lait (T.C.L.). Ann Dermatol Venereol 1980; 107: 313–327.
- Abeliovich D, Gelman-Kohan Z, Silverstein S, Lerer I, Chemke J, Merin S, et al. Familial café au lait spots: a variant of neurofibromatosis type 1. J Med Genet 1995; 32: 985–986.
- Viskochil D. Genetics of neurofibromatosis 1 and the NF1 gene. J Child Neurol 2002; 17: 562–651.
- Legius E, Brems H. Genetic basis of neurofibromatosis type 1 and related conditions, including mosaicism. Childs Nerv Syst 2020; 36: 2285–2295.
- Farschtschi S, Mautner VF, McLean ACL, Schulz A, Friedrich RE, Rosahl SK. The Neurofibromatoses. Dtsch Arztebl Int 2020; 117: 354–360.
- Tsipi M, Poulou M, Fylaktou I, Kosma K, Tsoutsou E, Pons MR, et al. Phenotypic expression of a spectrum of neurofibromatosis type 1 (NF1) mutations identified through NGS and MLPA. J Neurol Sci 2018; 395: 95–105.
- Frenk E, Marazzi A. Neurofibromatosis of von Recklinghausen: a quantitative study of the epidermal keratinocyte and melanocyte populations. J Invest Dermatol 1984; 83: 23–25.
- De Schepper S, Maertens O, Callens T, Naeyaert JM, Lambert J, Messiaen L. Somatic mutation analysis in NF1 café au lait spots reveals two NF1 hits in the melanocytes. J Invest Dermatol 2008; 128: 1050–1053.
- Boyd KP, Gao L, Feng R, Beasley M, Messiaen L, Korf BR, et al. Phenotypic variability among café-au-lait macules in neurofibromatosis type 1. J Am Acad Dermatol 2010; 63: 440–447.
- Kokkinou E, Roka K, Alexopoulos A, Tsina E, Nikas I, Krallis P, et al. Development of a multidisciplinary clinic of neurofibromatosis type 1 and other neurocutaneous disorders in Greece. A 3-year experience. Postgrad Med 2019; 131: 445–452.
- Taylor SC. Skin of color: biology, structure, function, and implications for dermatologic disease. J Am Acad Dermatol 2002; 46: S41–S62.
- Albaghdadi M, Thibodeau ML, Lara-Corrales I. Updated approach to patients with multiple café au lait macules. Dermatol Clin 2022; 40: 9–23.
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol 2009; 61: 1–16.
- Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH. Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol 2014; 75: 309–316.
- Pasmant E, Vidaud M, Vidaud D, Wolkenstein P. Neurofibromatosis type 1: from genotype to phenotype. J Med Genet 2012; 49: 483–489.
- Koczkowska M, Chen Y, Callens T, Gomes A, Sharp A, Johnson S, et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844–848. Am J Hum Genet 2018; 102: 69–87.
- Kehrer-Sawatzki H, Mautner VF, Cooper DN. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet 2017; 136: 349–376.
- Kang E, Kim YM, Seo GH, Oh A, Yoon HM, Ra YS, et al. Phenotype categorization of neurofibromatosis type I and correlation to NF1 mutation types. J Hum Genet 2020; 65: 79–89.
- Pemov A, Li H, Patidar R, Hansen NF, Sindiri S, Hartley SW, et al. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene 2017; 36: 3168–3177.
- De Schepper S, Boucneau J, Lambert J, Messiaen L, Naeyaert JM. Pigment cell-related manifestations in neurofibromatosis type 1: an overview. Pigment Cell Res 2005; 18: 13–24.
- Stamatas GN, Zmudzka BZ, Kollias N, Beer JZ. Non-invasive measurements of skin pigmentation in situ. Pigment Cell Res 2004; 17: 618–626.