Darier disease is a severe, rare autosomal dominant inherited skin condition caused by mutations in the ATP2A2 gene encoding sarcoendoplasmic reticulum Ca2+-ATPase isoform 2 in the endoplasmic reticulum. Since sarcoendoplasmic reticulum Ca2+-ATPase isoform 2 is expressed in most tissues, and intracellular calcium homeostasis is of fundamental importance, it is conceivable that other organs besides the skin may be involved in Darier disease. This review focusses on the association of Darier disease with other organ dysfunctions and diseases, emphasizing their common molecular pathology. In conclusion, Darier disease should be considered a systemic condition that requires systemic and disease mechanism targeted treatments.
Key words: Darier disease; genodermatosis; rare disease; endoplasmic reticulum; calcium; SERCA2.
Accepted Feb 16, 2021; Epub ahead of print Feb 19, 2021
Acta Derm Venereol 2021; 101: adv00430.
doi: 10.2340/00015555-3770
Corr: Etty Bachar-Wikström and Jakob D. Wikström, Dermato-Venereology Clinic, Karolinska University Hospital, SE-171 64 Stockholm, Sweden. E-mails: ester.bachar-wikstrom@ki.se; jakob.wikstrom@ki.se
SIGNIFICANCE
While research on the systemic aspects of Darier disease is still in its infancy, it is becoming clear that Darier disease is a multi-organ systemic condition. This not surprising, considering the overwhelming experimental evidence of the importance of sarcoendoplasmic reticulum Ca2+-ATPase isoform 2 in physiology and pathophysiology. While more data is needed, treating physicians should be aware of the risk of extracutaneous manifestations of Darier disease. Future research would benefit from more registry studies, as well as from systemic large-scale Darier disease cohort studies, in order to examine associations as well as to find direct experimental evidence of other organ dysfunctions and diseases.
INTRODUCTION
Darier (Darier-White) disease (DD), also known as keratosis follicularis, is an autosomal dominant inherited skin disease, which is thought to be caused by mutations in the ATP2A2 gene encoding sarcoendoplasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) (1). The disease was described independently by Darier and White in 1889, and the estimated prevalence ranges from 1 in 30,000 people in Scotland and 1 in 36,000 people in northern England, to 1 in 100,000 people in Scandinavia (2–4).
Since cellular calcium homeostasis is of fundamental importance, and the causative mutations are endogenously expressed, it is not surprising that a growing body of evidence indicates that DD is associated with non-dermatological conditions, such as epilepsy, neuropsychiatric disorders including mild intellectual disability, psychiatric disorders such as bipolar disorder and schizophrenia, as well as diabetes (5) and heart failure (6). This paper reviews DD and its association with the skin and other organs, emphasizing the connection between the molecular pathology that underlies these associated common conditions and DD.
GENETIC CAUSE
Mutations in the ATP2A2 gene were shown to be associated with DD in a landmark study in 1999 by Hovnanian et al. (1), which examined 13 families and sporadic cases. In an earlier publication, Parfitt et al. (7) mapped the disease genes to chromosome 12q23-q24.1, and the specific loci, which are between D12S78 and D12S79. The inheritance of one faulty copy of the gene causes a decrease in transcript production, which causes disease (haploinsufficiency) (8). Subsequently, more than 270 unique ATP2A2 mutations that disrupt critical functional domains of SERCA2 have been associated with DD (8, 9). These mutations vary between families and do not show gene clustering meaning (10). Most mutations described are missense mutations, in-frame deletions or insertions (63%) (http://www.hgmd.org). Other mutations will cause premature termination codons (PTC) or aberrant splicing (37%). Several studies aimed at identifying mutations leading to SERCA2 dysfunction or reduced expression, used a PCR amplification of exons and adjacent intronic splice sites of ATP2A2 (1, 11, 12). Thus, intronic mutations could have been missed. In an approach with similar coverage, Leong et al. used whole exome sequencing (WES) to screen the ATP2A2 gene in patients with DD, and identified 15 novel variants that were not previously reported in DD; however, when genotype-phenotype association was examined for all variants in relation to the patient’s disease severity score, no correlation could be established (13, 14). On the other hand, missense mutations were reported to be associated with a more severe disease phenotype elsewhere (1). Of note, out of 28 patients examined by Leong et al. (13), 4 patients did not have any variants in the ATP2A2 gene despite being clinically diagnosed with DD, and 2 patients had benign ATP2A2 variants. Although examined DD cohorts are small, the percentage of detected mutations is similar; Ringpfeil et al. (15) (58%), Onozuka et al. (16) (70%), and Leong et al. (13) (57%) . Thus, while the majority of DD cases appear to associate with ATP2A2, it cannot be excluded that undetected intronic mutations or deletions in this or even other genes could play a role in a subset of patients. Whole genome sequencing of DD cohorts would be needed to study this further.
SKIN INVOLVEMENT IN DARIER DISEASE
Skin symptoms and clinical features
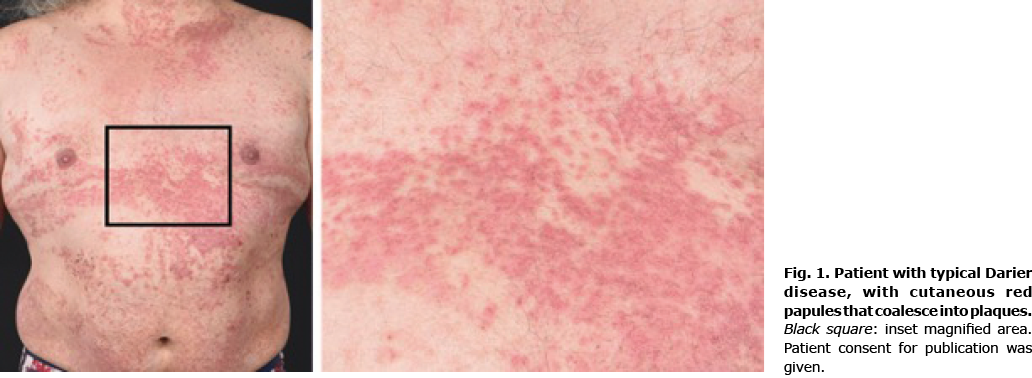
DD is characterized primarily by malodorous warty, greasy, yellow to brown, hyperkeratotic papules, on the seborrheic areas of the chest, upper back, forehead, scalp, nasolabial folds, and ears (Fig. 1) (2, 17). These lesions can lead to large crusted plaques with a high risk of acquiring secondary infections, particularly in folded areas (18). Typical nail abnormalities are characterized by longitudinal white or red lines with ridges and distal v-shaped notches on the nail surface (19, 20). In addition, papules may appear on mucosal membranes, mainly oral, pharynx, vulva and rectum (18). The whitish oral mucosal lesions mostly affect the hard palate and resemble nicotinic stomatitis (21). The gingivae is also a very commonly affected intraoral site, and when the disease affects the tongue it gives rise to a coarse, nodular appearance to the dorsum (22). In the majority of cases, the disease develops in adolescents or young adults and continues throughout life. Ultraviolet B (UVB) irradiation, heat, friction, and infections of affected areas are clinically known to exacerbate symptoms. The impact of DD goes beyond physical symptoms as shown by a severe reduction in health-related quality of life, which correlates with various disease characteristics, such as the skin area involved (23). The treatment of DD can be challenging and is often unsatisfactory (19). Currently, the most effective treatment is systemic retinoids, which may reduce hyperkeratosis, although the primary disease mechanism is not targeted.
Histopathological features
Microscopic examination of DD lesions reveals hyperkeratosis and hypergranulosis along with central keratin plugs. The majority of DD biopsies exhibit suprabasal clefts with acantholysis that extends through all levels of the epidermis with papillated epidermal hyperplasia (24–26). Examination of the epithelium reveals varying numbers of dyskeratotic cells in the stratum spinosum and stratum granulosum, described as “corps ronds” (round bodies) and parakeratotic cells in the stratum corneum which resemble “corps grains”. The underlying dermal connective tissue is fibrocellular and with mild chronic inflammation (25, 27), although in a case report neither neutrophilic nor eosinophilic infiltrates were observed (24). DD diagnosis is still primarily made with histopathology; however, it is expected that genetic tests will complement or replace this.
Keratinocyte adhesion
The epidermis of the skin is a thin, yet strong, protective layer. One of the classical histopathological signs of DD in skin biopsies is reduction in epidermal cell adhesion, acantholysis, mediated by a rupture of the desmosome-keratin complex (28). Desmosomes as well as adherens junctions are 2 types of cell-cell junctions with specific anchorage to keratinocyte intracellular intermediate or actin filaments that allow strong adhesion between cells and provide the epidermis with strength and resistance to mechanical stress (29, 30). The importance of Ca2+ homeostasis for the integrity and function of the epidermis has been studied extensively. For example, Duden & Franke (31) showed that keratinocytes cultured in media with low concentrations of Ca2+ show desmosomes disassembly, while physiological Ca2+ concentrations in the media direct desmosomal proteins to the plasma membrane where they form mature desmosomes (32). In DD patient-derived keratinocytes, as well as in keratinocytes treated with pharmacological inhibitors of the SERCA pumps, impairment of trafficking and distribution of desmosome components including desmoplakins, desmoglein, and desmocollin was found (33, 34). This may be due to extracellular increase in Ca2+, as this was shown to trigger transient desmosomal remodelling (35), or ER stress, as the molecular chaperone miglustat rescued ER stress impairment of cell-cell adhesion (36), although these are probably connected as ER stress is often associated with altered Ca2+ homeostasis. Li et al. (37) showed that, in human keratinocytes, pharmacological inhibition of SERCA impairs post ER maturation of desmosomal cadherins (DC), which, in patients with DD are mislocalized, further demonstrating that ER Ca2+ homeostasis is crucial to the ER to Golgi transport of nascent DC and that its impairment compromises keratinocyte adhesion. Similar results were shown in a canine kidney cell model for intercellular junction assembly, where inhibition of SERCA led to attenuating the formation of tight junctions and desmosome connections (38). It would be interesting to examine whether dysfunction in other ER Ca2+ regulators besides SERCA2 could create DD-like phenotypes.
Dyskeratinization and calcium dyshomeostasis
The epidermis continuously self-renews as keratinocytes grow outwards and differentiate. The gradient of calcium across the epidermis plays a crucial role in the keratinocyte differentiation, which is evident by simple cell culture experiments in which keratinocytes switch from proliferation to differentiation when cultured in high [Ca2+] (39). Recent studies implicate the ER as the major Ca2+ storage compartment, which forms the epidermal Ca2+ gradient (40–43). Indeed, histopathological examination of biopsies from patients with DD show focal dyskeratosis (premature differentiation of single keratinocytes) and hyperkeratosis (28). The round dyskeratotic cells termed “corps ronds” in the stratum spinosum and “grains” in the upper layers of the epidermis are probably apoptotic keratinocytes (28, 44). Leinonen et al. found lower epidermal Ca2+ in DD lesional compared with non-lesional skin (45); probably, however, not shown due to less SERCA2 expression and perhaps also due to skin barrier disruption, as this leads to ER Ca2+ release to the extracellular space (35).
Importantly, the majority of ATP2A2 mutations associated with DD exhibit a reduced expression and activity of the SERCA2 Ca2+ pump (46–50), therefore, an imbalance in cellular Ca2+ signalling is a hallmark in DD. SERCA2 is a 110-kDa transmembrane protein located in the ER, which is responsible for transporting Ca2+ back into the lumen of the ER from the cytosol (51) (Fig. 2). The overall structure is comprised of 3 cytoplasmic domains: an actuator, a nucleotide-binding, and a phosphorylation domain, as well as 10 transmembrane helices (52). Several studies show that SERCA is activated after binding of 2 Ca2+ ions, a process, which is ATP dependent and forms a phosphoenzyme, followed by release of Ca2+ ions into the ER/SR (53). Over the years, several studies, both in humans and in a murine model of the disease (heterozygous mice), suggested possible mechanisms leading to SERCA2 dysfunction. Ahn et al. (49) showed that most mutations in SERCA2b (out of 12 tested in this study) markedly affected protein expression; partially because of enhanced proteasomal degradation of mutant protein. Moreover, there was also a dominant negative effect of mutants over wild type (WT) pumps (49). In a murine DD model, Zhao et al. (54) showed that amylase exocytosis was ≈10-fold more sensitive to Ca2+ in cells from SERCA2+/– mice compared with WT-derived cells, which suggests a plasticity and adaptability of Ca2+ signalling and Ca2+-dependent cellular functions in vivo, which can explain the relatively normal function of most cells in patients with DD and that heterozygosity is compatible with life. Important to note is that SERCA2, specifically the SERCA2b isoform, is the SERCA type with the highest Km (55) and thus the highest Ca2+ pumping capacity, which may explain the vast impact of its mutations and why mutations in SERCA1 or SERCA3 have not been implicated in DD. SERCA pumps are encoded by 3 genes in humans (ATP2A1-3) that generate multiple isoforms (SERCAla,b, SERCA2a-c, SECA3a-f) by developmental or tissue-specific alternative splicing (56). These pumps differ by their regulatory and kinetic properties, allowing for optimized function in the tissue where they are expressed.

Endoplasmic stress
ER stress is defined as an imbalance between the protein load and the folding capacity of the ER, which results in accumulation of misfolded proteins, further activating an adaptive response termed the unfolded protein response (UPR). In mammals, The UPR response is carried out by 3 ER stress transducers, namely PKR-like endoplasmic reticulum kinase (PERK), inositol-requiring 1α (IRE1α), and activating transcription factor 6 (ATF6). The homeostatic impact of the UPR is played out at several levels (57). First, cells focus on translation attenuation to reduce the ER protein load. Secondly, the UPR upregulates the folding machinery by inducing ER chaperone genes. Thirdly, the ER compartment expands to accommodate the high protein load, and then activates ER-associated degradation (ERAD) of unfolded or misfolded proteins. However, if the UPR fails to resolve prolonged ER stress, apoptotic signals appear and cell death occurs. Thus, there exists a therapeutic window for balancing ER stress before tissue damage occurs.
ER stress is usually associated with ER calcium dyshomeostasis and the SERCA inhibitor thapsigargin is widely used to elicit ER stress experimentally. An immunohistochemical analysis of several ER stress markers in skin lesions derived from a patient with DD suggested a role of ER stress in the pathogenesis of DD (16). A vast genetic analysis of diverse mutants identical to those found in patients with DD demonstrated that SERCA2 mutated protein itself can initiate ER stress (51). These SERCA2 mutated proteins were found to be less soluble, to aggregate, and to be more polyubiquitinylated. After transduction into primary human keratinocytes, mutated SERCA2 aggregates elicited ER stress, led to increased numbers of cells to round up and detach from the culture plate, and induced apoptosis (51). Also, keratinocytes isolated from patients with DD exhibit prolonged ER stress, specifically, increased phosphorylation of eIF2α and IRE1α, which further contributes to aberrant cell-cell adhesion and keratinization and stimulates apoptosis (33, 58), while treatment with the drug miglustat restores adherens junctions and desmosomes in DD keratinocytes (33). One suggested explanation to the miglustat rescue effect is its action as a chaperone that allows the adhesion molecules to evade the UPR, reach the plasma membrane and form adhesion junctions and desmosomes. Another explanation offered by the authors is that miglustat, which is used clinically for type I Gaucher disease, also inhibits glucosylceramide synthase (59) and thus may act through its modulation of the ceramide/sphingolipid pathway (60).
Although speculative, it may be that ER stress induces preterm keratinocyte apoptosis that is identified histopathologically as corps rounds.
DARIER DISEASE AND DIABETES
Diabetes types
Diabetes refers to a group of metabolic diseases characterized by hyperglycaemia resulting from defects of insulin secretion and/ or increased resistance to insulin. Type 1 diabetes is developed following the destruction of β-cells in the pancreas, which leads to absolute insulin deficiency and full dependency on exogenous insulin for treatment. There are 2 main forms of type 1 diabetes; one is an immune-mediated disease with autoimmune markers, while the second form of type 1 diabetes named idiopathic diabetes, has no known cause and only a minority of patients fall into this group. Type 2 diabetes is by far the most common form of diabetes. In type 2 diabetes, the response to insulin is diminished, defined as insulin resistance (61). The disease is most commonly seen in people older than 45 years, but is increasingly seen in children, adolescents, and younger adults due to rising levels of obesity, physical inactivity, and high-calorie diets (62, 63).
Insulin secretion
In the pancreatic β-cell, glucose is acting as the major stimulator of insulin release. When the concentration of glucose increases, β-cell metabolism accelerates, leading to an increase in ATP/ADP ratio, which induces closure of KATP channels (64, 65). The resulting decrease in K+ efflux causes plasma membrane depolarization, followed by opening of Ca2+ channels and Ca2+ influx into the cell. The subsequent increase in cytosolic Ca2+ concentrations then promotes exocytosis of insulin granules (66, 67). The triggering Ca2+ signal is crucial. Experimental conditions that hamper the increase in cytosolic Ca2+ concentrations impair glucose-induced insulin secretion, while physiological or pharmacological means that increased β-cell cytosolic Ca2+ concentrations induce insulin secretion (68, 69).
In β-cells, as well as other cell types, there are 2 main Ca2+ release channels in the ER; the ryanodine receptor (RyR) and the inositol trisphosphate receptor (IP3R), which controls Ca2+ efflux (70, 71). SERCA2b and SERCA3a are the main SERCA isoforms found in the pancreatic β-cells (72) and control ER Ca2+ influx. These pumps ensure glucose-stimulated insulin release from the β-cell by maintaining high Ca2+ levels in the ER via pumping Ca2+ from the cytoplasm into ER. The involvement of SERCA2b/3a, as well as reduced RyR expression or function, in the pathogenesis of type 2 diabetes was reported (31, 73, 74). Furthermore, the role of ER stress in diabetes development has been massively investigated due to the fact that pancreatic β-cells are exposed to marked changes of ER protein load in response to daily physiological changes in insulin demand. The pancreatic β-cells are therefore, highly susceptible to ER stress under conditions of increased insulin production, as occurs during prolonged hyperglycaemia (75, 76). Chronic ER stress associated with diabetes can impair protein folding in the ER, reduce insulin secretion, induce oxidative stress, and lead to β-cell death (77). In this context, ER stress might occur due to pathological, environmental, and genetic factors including glucolipotoxicity, inflammatory responses, amyloid accumulation, and expression of mutant proinsulin (78–81). The indication that ER stress may play a role in the pathogenesis of autoimmune diabetes came from recent reports that demonstrate the presence of some of the ER stress markers in inflamed islets of both diabetes-prone non-obese diabetic (NOD) mice (82) and patients with autoimmune diabetes (83). Engin et al. (84) showed diminished expression of the UPR markers spliced XBP-1 and ATF6 in β-cells of NOD mice and human patients during the disease progression. This study emphasizes the delicate balance of the unfolded protein response to ER stress, as was demonstrated previously (85). A continuous diabetic environment might lead to prolonged ER stress overcoming the cell capability to reduce ER stress by failing to activate UPR branches. In this case, a failed adaptive UPR response can correlate with β-cell death and insulin deficiency in both experimental models and human T1D (84).
Due to the importance of ER calcium homeostasis for β-cell physiology and pathophysiology, Cederlöf et al. (86) examined the potential association of DD with diabetes at the population level. Individuals with DD had a 74% increased risk of being diagnosed with type 1 diabetes; however, no increased risk of type 2 diabetes was found. This study was performed in a Swedish population, and more data acquired from other populations will be needed to verify DD as a risk factor for type 1 diabetes. Several monogenic forms of diabetes provide strong evidence for the crucial role of single molecules in causing diabetes in humans. For example, INS1 mutation or the Akita insulin mutation (the later initially described in mice (87)), will lead to neonatal diabetes (88), maturity onset diabetes of the young (MODY) or antibody negative T1D. In addition, mutations in EIF2AK3 (part of the PERK signalling arm of the UPR) cause Wollcot-Rallison syndrome (89), and recessive mutations in WFS1, encoding the ER resident protein wolframin 1 that regulates calcium and ER protein folding, leads to Wolfram syndrome 1. Of note, some dominant mutations in WFS1 can also cause Wolfram syndrome-like disease, which can appear with or without diabetes (90–92). Both of these autosomal recessive disorders show ER stress and cause young-onset diabetes (93). In accordance with this, several studies show the importance of proper functioning SERCA2 in preventing β-cell death and diabetes in response to ER stress (35, 94, 95). A recent study supports an important role of SERCA2 in β-cells in humans, as patients with DD have an altered β-cell phenotype (5). Specifically, evidence of basal hyper-insulin secretion was found by the Homeostasis Model Assessment HOMA2-%, a marker for basal insulin secretion (5). It is notable that many more genes, mainly Human Leukocyte Antigen (HLA)-related, are linked to different forms of diabetes, as described in detail by Yang et al. (96).
What are the possible molecular causes behind type 1 diabetes risk in DD? Several of the autoantigens in autoimmune diseases, such as coeliac disease (97), collagen-induced arthritis (98), multiple sclerosis/experimental autoimmune encephalomyelitis (99), rheumatoid arthritis (100), and systemic lupus erythematosus (101) undergo post-translational modifications in the ER, which is important for the proteins’ normal function, as well as antigen properties. ER stress per se can also activate enzymes that alter post-translational modifications of proteins and thus create neoautoantigens, which have a pathological role in type 1 diabetes (102). Likewise, some of the environmental triggers for type 1 diabetes, such as Coxsackie viral infection, have been shown to increase β-cell ER stress, which may increase production of autoantigens or create neo-autoantigens (103). Autoantigens are sensed by dendritic cells engulfing apoptotic β-cells and stimulate the maturation of β-cell reactive T-cells. Hence, abnormal ER stress may stimulate immuno-destruction of β-cells. DD and type 1 diabetes typically both debut in adolescence, which suggests a possible common pathophysiology. While speculative, it may be that ER stress in β-cells in patients with DD interferes with normal protein folding and thus creates neoautoantigens in a similar manner, which induce regular autoimmune β-cell destruction.
DARIER DISEASE AND HEART FAILURE
Cardiomyocyte calcium homeostasis
The 2 major SERCA2 protein isoforms are the housekeeping SERCA2b, which is expressed in all tissues at low levels (104), and the more specialized SERCA2a isoform, predominantly expressed in cardiac and slow-twitch skeletal muscle (105). In the heart, a raised intracellular Ca2+ concentration is the trigger that activates cardiomyocyte contraction. Specifically, Ca2+ enters through the L-type channels, located primarily at sarcoplasmic reticulum (SR) junctions. The influx of Ca2+ triggers the release of further Ca2+ from the SR via ryanodine receptor 2 (RyR2). Elevated free intracellular Ca2+ will result in interaction between actin and myosin, shortening of sarcomeres and contraction. Diastolic relaxation is an active (ATP-dependent) process, in which Ca2+ transport out of the cytosol occurs via the SR Ca2+ ATPase SERCA2a, leading to a decrease in intracellular Ca2+ concentrations required for both muscle relaxation (106) and for replenishing Ca2+ stores needed for the next contraction (107).
It is widely described that deranged cardiomyocyte Ca2+ kinetics can cause heart failure (108). Heart failure (HF) is a major public health problem, associated with significant mortality, morbidity, and healthcare expenses, particularly among those above the age of 65 years (109, 110). SERCA2a expression is downregulated in heart failure (111–114). Consequently, in vivo gene transfer of SERCA2a in pigs preserved systolic function and improved ventricular remodelling (115), whereas restoring SERCA2a expression by gene transfer in heart-failure patients showed some contradicting results (116, 117). Thus, more research is needed into the significance of SERCA2 in human heart failure.
An important common pathological event between HF and DD is ER stress. As mentioned earlier, depletion of ER luminal Ca2+ may induce the UPR. UPR-induced apoptosis has been implicated in the pathophysiology of heart failure, as well as in other cardiovascular diseases (118–123). Specifically, patients with HF display structural and architecture alteration of the ER, as well as maladaptation of the ER proteins involved in the UPR (124). Several ER stress markers, such as spliced X-box binding protein 1 (XBP1s), glucose-regulated protein 78 (GRP78), activating transcription factor 4 (ATF4), and C/EBP-homologous protein (CHOP) were all shown to be induced in HF in humans (118, 124–127). In conjunction with the human data, animal studies, mainly in mice, showed a protective role of ER stress in HF; hearts of PERK knockout mice showed a significant reduction in Serca2α expression, an increase in apoptosis and UPR genes expression (GRP78, GRP94, CHOP) in response to induction of heart failure (128), and thrombospondin (Thbs) knockout mice showed reduced activation of Atf6α with injury and Thbs4-mediated protection was lost upon Atf6α deletion (129). Overall, the involvement of ER stress in HF is well established, however, one should bear in mind the differences between the models studied and the implications for humans.
Since DD and HF display a common pathological mechanism due to perturbation in SERCA2, the susceptibility of patients with DD to HF was further examined. Interestingly, SERCA2 haplosufficient mice were reported to develop HF when crossed with a transgenic model of increased myofibrillar Ca2+-sensitivity, suggesting that patients with DD might be more susceptible to HF (8). Indeed, in a recent publication, it was shown that patients with DD exhibit a disease-specific increased risk of HF at an earlier age compared with healthy controls (6). Interestingly, female patients with DD showed a higher risk for heart disease in general. This observation contributes to the evidence of an important role of SERCA2 in human HF pathophysiology and suggests that perhaps HF in patients with DD is a specific subtype that requires tailored treatment. More studies are needed to corroborate DD as a risk factor for HF.
DARIER DISEASE AND THE BRAIN
The idea that patients with skin conditions (e.g. psoriasis, eczema, and skin cancer) frequently face psychological challenges, such as depression, anxiety and suicidality and overall poor quality of life (130–132) is well known. This is attributed to the stress related to the avoidance-coping mechanism, as well as social and activity prevention. However, in the case of patients with DD, the link between the severe skin condition and psychological problems is likely to be of a genotype-phenotype correlation in many cases and, in fact, DD-causing mutations in ATP2A2 increase the susceptibility to neuropsychiatric dysfunction, in particular severe psychiatric illness (133). The mechanism underlying this correlation probably lies in the important role of Ca2+ in brain function. Indeed, disruption of normal Ca2+ flux in the brain has been associated with psychiatric conditions, including bipolar disorder, schizophrenia, autism spectrum disorders, and intellectual disabilities (134, 135), as well as neurodegenerative disorders, such as Alzheimer’s disease (136, 137) and Parkinson’s disease (138). As in the case of heart and skin, in the neuronal system, Ca2+ released into the cytoplasm is pumped back into the ER by SERCA2 (139). SERCA2a expression has been identified in the brain at low levels, and is found in the granular cells of cerebellar Purkinje neurones, as well as in the giant cells of the brainstem reticular formation (140, 141). SERCA2b is expressed at higher levels in neuronal microsomes, synaptic plasma membrane vesicles, and synaptosomes (142). An early association between SERCA2 and neuropsychiatric pathophysiology was made in patients with DD by Jacobsen et al. (135) and by Craddock et al. (143), and additional work has shown that patients with DD have an increased prevalence of several neuropsychiatric disorders, including depression (30%), bipolar disorder (4%), epilepsy (3%), schizophrenia (1%) and cognitive disabilities (4%) (31, 144–148). One possible explanation for common effects of mutations in the SERCA2 gene on skin and brain is that these tissues share a common ectodermal origin and thus may share sensitivity towards SERCA2 impairment (149). Interestingly, the drug lithium, which is used to treat bipolar disease and acts to alter intraneuronal Ca2+ concentrations, distribution and signalling (150, 151), was reported to exacerbate or even cause DD (152, 153) and, indeed, lithium was shown to decrease SERCA2 expression in rats (154). It would certainly be interesting to examine whether specific ER Ca2+ targeting treatments would impact the different neuropsychatric conditions associated with DD. While the neuropsychiatric association of DD is well explored, further research is needed to examine whether DD is associated with neurodegenerative conditions.
DARIER DISEASE AND CANCER
It is not known whether DD is associated with cancer; however, several in vitro and animal studies imply that this is worth examining. An ageing study in heterozygous Atp2a2–/+ mice revealed a higher incidence of squamous cell carcinoma of the oral mucosa, i.e. of keratinocytes, which are also affected in human DD (155). Human-derived primary oral tumours showed that the ATP2A2 gene is downregulated, which may be partly regulated by an epigenetic mechanism (156). Moreover, downregulation of both SERCA2 and SERCA3 expression was reported in thyroid and colon cancer cell lines (157, 158). Korosec et al. (159) showed that germline alterations of ATP2A2 in humans may predispose to lung and colon cancer, and that an impaired ATP2A2 gene might be involved as an early event in carcinogenesis. Mechanistically, studies in mice showed that Atp2a2 haploinsufficiency dramatically affects keratinocytes overall gene expression and differentiation, leading to alteration of the tissue environment, which may be permissive for tumour development (160, 161). In addition, biopsies from patients with colorectal cancer showed high expression of SERCA2 (162) and overexpression of SERCA2b in human colon adenocarcinoma (162), and liposarcoma (163) promoted cell proliferation and migration. Thus, SERCA2 mutations, such as in DD, may affect oncogenesis at several levels. Although speculative, it may be that DD is associated with increased risk for some malignancies, and reduced risk for others.
CURRENT AND FUTURE TREATMENTS
As for many other rare diseases, a long list of suggested treatments usually indicates that no one treatment works well. Treatments for mild symptoms of DD include moisturizers, sun protection, and careful selection of clothing to avoid heat and sweating (20). Localized DD lesions can be treated with topical glucocorticosteroids or vitamin D3 ointment (164), mainly aiming to reduce inflammation without altering the disease course (165). Several topical retinoids, such as isotretinoin (166), tazarotene (167), and adapalene (168), were also reported as DD treatments. Topical retinoids and retinoid analogues help normalize cell turnover and cell cohesion (169), with isotretinoin also exhibiting a strong inhibition of cell proliferation (170, 171). Ultimately these actions facilitate the levelling of papules and reduce hyperkeratosis. It is not known if retinoids have any effect on keratinocyte calcium homeostasis, thus targeting the specific DD pathology, although in ovarian follicle granulosa cells retinoic acid was shown to increase ER stores of Ca2+ (172), and in myelogenous and promyelocytic bone marrow cells retinoic receptor activation was associated with reduced ER stores of Ca2+ (173). Moreover, in vitro studies have demonstrated an anti-inflammatory effect of retinoids, which might be of additional benefit in DD, since the skin lesions can become inflamed (174, 175). For effective removal of DD refractory-proliferative lesions surgical approaches, such as dermabrasion, electrodessication, and various ablative laser modalities, have sometimes been used (176). In clinical routine, severe symptoms of DD are currently mainly treated systemically with the oral retinoids acitretin (177) or isotretinoin (178). Although showing efficacy on skin lesions (170), recurrence always occurs after ceasing treatment and adverse effects are common (170). These include dry lips, cheilitis, scaling, skin atrophy and fragility, as well as hepatotoxicity, ophthalmological complications, pancreatitis, and skeletal alterations (179). Consequently, patients might undergo intermittent treatment or even discontinuation of treatment (4). Of note, in rare cases DD has also been treated with the immunosuppressant cyclosporine A (180).
It is obvious that novel, disease mechanism targeted treatments are needed, and these should probably be systemic in order to target multiple organ dysfunctions. Moreover, perhaps these treatments should be commenced early in life in order to prevent, rather than just treat, the disease. As discussed earlier, impaired SERCA is at the core of DD. Pharmacological activation of SERCA may therefore ameliorate DD-related symptoms. A recent study (36) showed that CDN1163, a SERCA activator that directly binds to the SERCA2b enzyme to activate its Ca2+-ATPase activity probably via an allosteric mechanism (181), improved metabolic parameters in vivo in a type 2 diabetes mouse model (ob/ob) (36). Moreover, Savignac et al. (36) showed that ER stress and associated impairment of cell-to-cell adhesion in patients with DD-derived primary keratinocytes were rescued by the pharmacological chaperone miglustat, which is a clinically available drug. Thus, candidates that affect ER calcium or ER stress exist and could be included in clinical trials.
GENETIC COUNSELLING AND PATIENT INFORMATION
Since DD is usually inherited, a method of preventing the disease rather than just treating it is to perform preimplantation genetic diagnosis of fertilized embryos, in which 1 or 2 cells (blastomeres) are removed from the preimplantation embryo at the 6–10-cell stage (day 3 of development), thus allowing replacement into the uterus of unaffected embryos. This is, of course, a difficult ethical decision; however, as the systemic aspects of DD become more established more parents are likely to consider this alternative and have the right to be fully informed about it by their dermatologists and referred for genetic counselling (17). In our experience, most patients are unaware of this option. Moreover, it is important to perform genetic testing on all patients, as some do not have pathological mutations and, in our experience, some patients choose not to have children in order not to transmit the disease. While more studies are needed on the systemic aspects of DD, patients should be fully informed on the systemic aspects of their disease, although we cannot currently offer any treatments that target organs other than the skin.
CONCLUSION
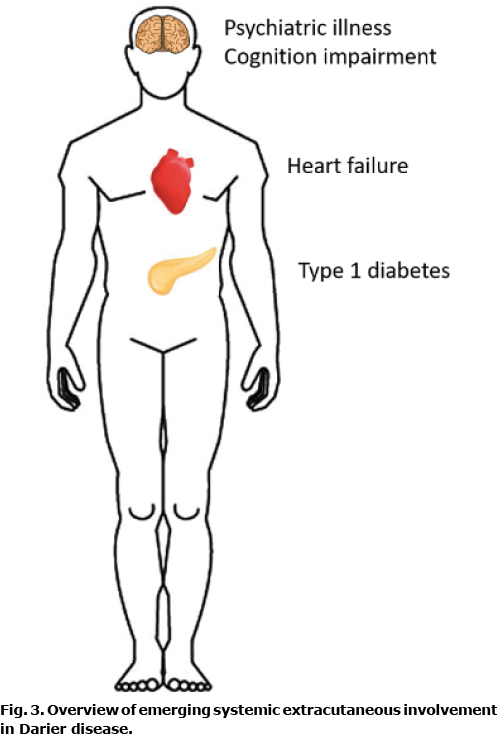
While research on the systemic aspects of DD is still in its infancy, it is becoming clear that DD is a multi-organ systemic condition (Fig. 3). This not surprising, considering the overwhelming experimental evidence of the importance of SERCA2 in physiology and pathophysiology. While more data is needed, treating physicians should be aware of the risk of extracutaneous manifestations. Future research would benefit from more registry studies, as well as from systemic large-scale DD cohort studies, in order to examine associations as well as to find direct experimental evidence of other organ dysfunctions and diseases.
ACKNOWLEDGEMENTS
The authors would like to extend sincere thanks and appreciation to the participating patient in Fig. 1. The patients in this manuscript have given written informed consent to publication. The authors are also grateful to the funding agencies (Vetenskapsrådet, Hudfonden, Svenska Sällskapet för medicinsk forskning, ALF medicin Stockholm, Jeanssons stiftelse, Tore Nilssons Stiftelse) for their support. Jakob Wikström was supported by a Wallenberg clinical fellow grant from the Marianne and Marcus Wallenberg foundation.
The authors have no conflicts of interest to declare.
REFERENCES
- Sakuntabhai A, Ruiz-Perez V, Carter S, Jacobsen N, Burge S, Monk S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet 1999; 21: 271–277.
- Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol 1992; 27: 40–50.
- Tavadia S, Mortimer E, Munro CS. Genetic epidemiology of Darier’s disease: a population study in the west of Scotland. Br J Dermatol 2002; 146: 107–109.
- Svendsen IB, Albrectsen B. The prevalence of dyskeratosis follicularis (Darier’s disease) in Denmark: an investigation of the heredity in 22 families. Acta Derm Venereol 1959; 39: 256–629.
- Ahanian T, Curman P, Leong IUS, Brismar K, Bachar-Wikstrom E, Cederlof M, et al. Metabolic phenotype in Darier disease: a cross-sectional clinical study. Diabetol Metab Syndr 2020; 12:12.
- Bachar-Wikstrom E, Curman P, Ahanian T, Leong IUS, Larsson H, Cederlof M, et al. Darier disease is associated with heart failure: a cross-sectional case-control and population based study. Sci Rep 2020; 10: 6886.
- Parfitt E, Burge S, Craddock N, Roberts E, McLean WH, Weissenbach J, et al. The gene for Darier’s disease maps between D12S78 and D12S79. Hum Mol Genet 1994; 3: 35–38.
- Prasad V, Lorenz JN, Lasko VM, Nieman ML, Huang W, Wang Y, et al. SERCA2 haploinsufficiency in a mouse model of Darier disease causes a selective predisposition to heart failure. Biomed Res Int 2015; 2015: 251598.
- Nellen RG, Steijlen PM, van Steensel MA, Vreeburg M, European Professional Contributors; Frank J, et al. Mendelian disorders of cornification caused by defects in intracellular calcium pumps: mutation update and database for variants in ATP2A2 and ATP2C1 associated with Darier Disease and Hailey-Hailey disease. Hum Mutat 2017; 38: 343–356.
- Elizondo LI, Jafar-Nejad P, Clewing JM, Boerkoel CF. Gene clusters, molecular evolution and disease: a speculation. Curr Genomics 2009; 10: 64–75.
- Sakuntabhai A, Dhitavat J, Burge S, Hovnanian A. Mosaicism for ATP2A2 mutations causes segmental Darier’s disease. J Invest Dermatol 2000; 115: 1144–1147.
- Yasuno S, Miyoshi Y, Asano N, Okita T, Yamaguchi M, Shimomura N, et al. Sporadic case of Darier disease caused by a novel splice-site mutation in the ATP2A2 gene. Clin Exp Dermatol 2019; 44: e10–e12.
- Leong IUS, Stuckey A, Ahanian T, Cederlof M, Wikstrom JD. Novel mutations in Darier disease and association to self-reported disease severity. PLoS One 2017; 12: e0186356.
- Noda K, Takeichi T, Okuno Y, Takama H, Miura S, Kagami S, et al. Novel and recurrent ATP2A2 mutations in Japanese patients with Darier’s disease. Nagoya J Med Sci 2016; 78: 485–492.
- Ringpfeil F, Raus A, DiGiovanna JJ, Korge B, Harth W, Mazzanti C, et al. Darier disease – novel mutations in ATP2A2 and genotype-phenotype correlation. Exp Dermatol 2001; 10: 19–27.
- Onozuka T, Sawamura D, Goto M, Yokota K, Shimizu H. Possible role of endoplasmic reticulum stress in the pathogenesis of Darier’s disease. J Dermatol Sci 2006; 41: 217–220.
- Hulatt L, Burge S. Darier’s disease: hopes and challenges. J R Soc Med 2003; 96: 439–441.
- Hovnanian A. Darier’s disease: from dyskeratosis to endoplasmic reticulum calcium ATPase deficiency. Biochem Biophys Res Commun 2004; 322: 1237–1244.
- Sehgal VN, Srivastava G. Darier’s (Darier-White) disease/keratosis follicularis. Int J Dermatol 2005; 44: 184–192.
- Cooper SM, Burge SM. Darier’s disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol 2003; 4: 97–105.
- Bernabe DG, Kawata LT, Beneti IM, Crivelini MM, Biasoli ER. Multiple white papules in the palate: oral manifestation of Darier’s disease. Clin Exp Dermatol 2009; 34: e270–e271.
- Macleod RI, Munro CS. The incidence and distribution of oral lesions in patients with Darier’s disease. Br Dent J 1991; 171: 133–136.
- Dodiuk-Gad R, Cohen-Barak E, Ziv M, Shani-Adir A, Amichai B, Zlotogorski A, et al. Health-related quality of life among Darier’s disease patients. J Eur Acad Dermatol Venereol 2013; 27: 51–56.
- See SHC, Peternel S, Adams D, North JP. Distinguishing histopathologic features of acantholytic dermatoses and the pattern of acantholytic hypergranulosis. J Cutan Pathol 2019; 46: 6–15.
- Kositkuljorn C, Suchonwanit P. Darier’s Disease: report of a case with facial involvement. Case Rep Dermatol 2019; 11: 327–333.
- Gupta LK, Garg A, Khare AK, Mittal A. A case of zosteriform Darier’s disease with seasonal recurrence. Indian Dermatol Online J 2013; 4: 219–221.
- Suryawanshi H, Dhobley A, Sharma A, Kumar P. Darier disease: a rare genodermatosis. J Oral Maxillofac Pathol 2017; 21: 321.
- Hakuno M, Shimizu H, Akiyama M, Amagai M, Wahl JK, Wheelock MJ, et al. Dissociation of intra- and extracellular domains of desmosomal cadherins and E-cadherin in Hailey-Hailey disease and Darier’s disease. Br J Dermatol 2000; 142: 702–711.
- Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol 2004; 5: 271–81.
- Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta 2008; 1778: 572–587.
- Duden R, Franke WW. Organization of desmosomal plaque proteins in cells growing at low calcium concentrations. J Cell Biol 1988; 107: 1049–1063.
- Pillai S, Bikle DD, Hincenbergs M, Elias PM. Biochemical and morphological characterization of growth and differentiation of normal human neonatal keratinocytes in a serum-free medium. J Cell Physiol 1988; 134: 229–237.
- Savignac M, Simon M, Edir A, Guibbal L, Hovnanian A. SERCA2 dysfunction in Darier disease causes endoplasmic reticulum stress and impaired cell-to-cell adhesion strength: rescue by Miglustat. J Invest Dermatol 2014; 134: 1961–1970.
- Dhitavat J, Cobbold C, Leslie N, Burge S, Hovnanian A. Impaired trafficking of the desmoplakins in cultured Darier’s disease keratinocytes. J Invest Dermatol 2003; 121: 1349–1355.
- Celli A, Crumrine D, Meyer JM, Mauro TM. Endoplasmic reticulum calcium regulates epidermal barrier response and desmosomal structure. J Invest Dermatol 2016; 136: 1840–1847.
- Kang S, Dahl R, Hsieh W, Shin A, Zsebo KM, Buettner C, et al. Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders. J Biol Chem 2016; 291: 5185–5198.
- Li N, Park M, Xiao S, Liu Z, Diaz LA. ER-to-Golgi blockade of nascent desmosomal cadherins in SERCA2-inhibited keratinocytes: Implications for Darier’s disease. Traffic 2017; 18: 232–241.
- Stuart RO, Sun A, Bush KT, Nigam SK. Dependence of epithelial intercellular junction biogenesis on thapsigargin-sensitive intracellular calcium stores. J Biol Chem 1996; 271: 13636–13641.
- Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab 2012; 7: 461–472.
- Menon GK, Grayson S, Elias PM. Ionic calcium reservoirs in mammalian epidermis: ultrastructural localization by ion-capture cytochemistry. J Invest Dermatol 1985; 84: 508–512.
- Forslind B. Quantitative X-ray microanalysis of skin. Particle probe evaluation of the skin barrier function. Acta Derm Venereol 1987; Suppl 134: 1–8.
- Lee SH, Elias PM, Proksch E, Menon GK, Mao-Quiang M, Feingold KR. Calcium and potassium are important regulators of barrier homeostasis in murine epidermis. J Clin Invest 1992; 89: 530–538.
- Menon GK, Elias PM, Lee SH, Feingold KR. Localization of calcium in murine epidermis following disruption and repair of the permeability barrier. Cell Tissue Res 1992; 270: 503–512.
- Godic A. Darier disease: a guide to the physician. J Med 2004; 35: 5–17.
- Leinonen PT, Hagg PM, Peltonen S, Jouhilahti EM, Melkko J, Korkiamaki T, et al. Reevaluation of the normal epidermal calcium gradient, and analysis of calcium levels and ATP receptors in Hailey-Hailey and Darier epidermis. J Invest Dermatol 2009; 129: 1379–1387.
- Dode L, Andersen JP, Leslie N, Dhitavat J, Vilsen B, Hovnanian A. Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 2 isoforms and characterization of Darier disease (SERCA2) mutants by steady-state and transient kinetic analyses. J Biol Chem 2003; 278: 47877–47889.
- Ikeda S, Mayuzumi N, Shigihara T, Epstein EH, Jr, Goldsmith LA, Ogawa H. Mutations in ATP2A2 in patients with Darier’s disease. J Invest Dermatol 2003; 121: 475–477.
- Sakuntabhai A, Burge S, Monk S, Hovnanian A. Spectrum of novel ATP2A2 mutations in patients with Darier’s disease. Hum Mol Genet 1999; 8: 1611–1619.
- Ahn W, Lee MG, Kim KH, Muallem S. Multiple effects of SERCA2b mutations associated with Darier’s disease. J Biol Chem 2003; 278: 20795–20801.
- Miyauchi Y, Daiho T, Yamasaki K, Takahashi H, Ishida-Yamamoto A, Danko S, et al. Comprehensive analysis of expression and function of 51 sarco(endo)plasmic reticulum Ca2+-ATPase mutants associated with Darier disease. J Biol Chem 2006; 281: 22882–22895.
- Wang Y, Bruce AT, Tu C, Ma K, Zeng L, Zheng P, et al. Protein aggregation of SERCA2 mutants associated with Darier disease elicits ER stress and apoptosis in keratinocytes. J Cell Sci 2011; 124: 3568–3580.
- Zhang Y, Inoue M, Tsutsumi A, Watanabe S, Nishizawa T, Nagata K, et al. Cryo-EM structures of SERCA2b reveal the mechanism of regulation by the luminal extension tail. Sci Adv 2020; 6: eabb0147.
- Lee AG. Ca2+ -ATPase structure in the E1 and E2 conformations: mechanism, helix-helix and helix-lipid interactions. Biochim Biophys Acta 2002; 1565: 246–266.
- Zhao XS, Shin DM, Liu LH, Shull GE, Muallem S. Plasticity and adaptation of Ca2+ signaling and Ca2+-dependent exocytosis in SERCA2(+/–) mice. EMBO J 2001; 20: 2680–2689.
- Vandecaetsbeek I, Trekels M, De Maeyer M, Ceulemans H, Lescrinier E, Raeymaekers L, et al. Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc Natl Acad Sci U S A 2009; 106: 18533–18538.
- Hovnanian A. SERCA pumps and human diseases. Subcell Biochem 2007; 45: 337–363.
- Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015; 11: 1956–1977.
- Mauro T. Endoplasmic reticulum calcium, stress, and cell-to-cell adhesion. J Invest Dermatol 2014; 134: 1800–1801.
- Venier RE, Igdoura SA. Miglustat as a therapeutic agent: prospects and caveats. J Med Genet 2012; 49: 591–597.
- Celli A, Mackenzie DS, Zhai Y, Tu CL, Bikle DD, Holleran WM, et al. SERCA2-controlled Ca(2)+-dependent keratinocyte adhesion and differentiation is mediated via the sphingolipid pathway: a therapeutic target for Darier’s disease. J Invest Dermatol 2012; 132: 1188–1195.
- Wilcox G. Insulin and insulin resistance. Clin Biochem Rev 2005; 26: 19–39.
- Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 2018; 14: 88–98.
- Eckstein ML, Williams DM, O’Neil LK, Hayes J, Stephens JW, Bracken RM. Physical exercise and non-insulin glucose-lowering therapies in the management of type 2 diabetes mellitus: a clinical review. Diabet Med 2019; 36: 349–358.
- Tarasov A, Dusonchet J, Ashcroft F. Metabolic regulation of the pancreatic beta-cell ATP-sensitive K+ channel: a pas de deux. Diabetes 2004; 53: S113–S122.
- Detimary P, Van den Berghe G, Henquin JC. Concentration dependence and time course of the effects of glucose on adenine and guanine nucleotides in mouse pancreatic islets. J Biol Chem 1996; 271: 20559–20565.
- Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem 1999; 259: 3–17.
- Rorsman P, Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003; 46: 1029–1045.
- Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 2000; 49: 1751–1760.
- Henquin JC. Pathways in beta-cell stimulus-secretion coupling as targets for therapeutic insulin secretagogues. Diabetes 2004; 53: S48–S58.
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev 2002; 82: 893–922.
- Mikoshiba K. Role of IP3 receptor signaling in cell functions and diseases. Adv Biol Regul 2015; 57: 217–227.
- Varadi A, Molnar E, Ostenson CG, Ashcroft SJ. Isoforms of endoplasmic reticulum Ca(2+)-ATPase are differentially expressed in normal and diabetic islets of Langerhans. Biochem J 1996; 319: 521–527.
- Roe MW, Philipson LH, Frangakis CJ, Kuznetsov A, Mertz RJ, Lancaster ME, et al. Defective glucose-dependent endoplasmic reticulum Ca2+ sequestration in diabetic mouse islets of Langerhans. J Biol Chem 1994; 269: 18279–18282.
- Islam MS. The ryanodine receptor calcium channel of beta-cells: molecular regulation and physiological significance. Diabetes 2002; 51: 1299–1309.
- Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS One 2008; 3: e1648.
- Leibowitz G, Bachar E, Shaked M, Sinai A, Ketzinel-Gilad M, Cerasi E, et al. Glucose regulation of beta-cell stress in type 2 diabetes. Diabetes Obes Metab 2010; 12: 66–75.
- Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev 2008; 29: 317–333.
- Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS One 2009; 4: e4954.
- Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 2010; 120: 744–755.
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol 2012; 197: 857–867.
- Bachar-Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, et al. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 2013; 62: 1227–1237.
- Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, et al. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012; 61: 818–827.
- Marhfour I, Lopez XM, Lefkaditis D, Salmon I, Allagnat F, Richardson SJ, et al. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012; 55: 2417–2420.
- Engin F, Yermalovich A, Nguyen T, Hummasti S, Fu W, Eizirik DL, et al. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci Transl Med 2013; 5: 211ra156.
- Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell 2009; 35: 551–561.
- Cederlof M, Curman P, Ahanian T, Leong IUS, Brismar K, Bachar-Wikstrom E, et al. Darier disease is associated with type 1 diabetes: findings from a population-based cohort study. J Am Acad Dermatol 2019; 81: 1425–1426.
- Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest 1999; 103: 27–37.
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A 2007; 104: 15040–15044.
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 2000; 25: 406–409.
- Eiberg H, Hansen L, Kjer B, Hansen T, Pedersen O, Bille M, et al. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet 2006; 43: 435–440.
- Rendtorff ND, Lodahl M, Boulahbel H, Johansen IR, Pandya A, Welch KO, et al. Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am J Med Genet A 2011; 155A: 1298–1313.
- Pallotta MT, Tascini G, Crispoldi R, Orabona C, Mondanelli G, Grohmann U, et al. Wolfram syndrome, a rare neurodegenerative disease: from pathogenesis to future treatment perspectives. J Transl Med 2019; 17: 238.
- Rigoli L, Di Bella C. Wolfram syndrome 1 and Wolfram syndrome 2. Curr Opin Pediatr 2012; 24: 512–517.
- Tong X, Kono T, Anderson-Baucum EK, Yamamoto W, Gilon P, Lebeche D, et al. SERCA2 deficiency impairs pancreatic beta-cell function in response to diet-induced obesity. Diabetes 2016; 65: 3039–3052.
- Kono T, Ahn G, Moss DR, Gann L, Zarain-Herzberg A, Nishiki Y, et al. PPAR-gamma activation restores pancreatic islet SERCA2 levels and prevents beta-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol Endocrinol 2012; 26: 257–271.
- Yang Y, Chan L. Monogenic diabetes: what it teaches us on the common forms of type 1 and type 2 diabetes. Endocr Rev 2016; 37: 190–222.
- Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med 1998; 4: 713–717.
- Corthay A, Backlund J, Broddefalk J, Michaelsson E, Goldschmidt TJ, Kihlberg J, et al. Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur J Immunol 1998; 28: 2580–2590.
- Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L, Rothbard JB. T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature 1986; 324: 258–260.
- Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest 1998; 101: 273–281.
- Mamula MJ, Gee RJ, Elliott JI, Sette A, Southwood S, Jones PJ, et al. Isoaspartyl post-translational modification triggers autoimmune responses to self-proteins. J Biol Chem 1999; 274: 22321–22327.
- Marre ML, Piganelli JD. Environmental factors contribute to beta cell endoplasmic reticulum stress and neo-antigen formation in type 1 diabetes. Front Endocrinol (Lausanne) 2017; 8: 262.
- van Kuppeveld FJ, Hoenderop JG, Smeets RL, Willems PH, Dijkman HB, Galama JM, et al. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J 1997; 16: 3519–3532.
- de la Bastie D, Levitsky D, Rappaport L, Mercadier JJ, Marotte F, Wisnewsky C, et al. Function of the sarcoplasmic reticulum and expression of its Ca2(+)-ATPase gene in pressure overload-induced cardiac hypertrophy in the rat. Circ Res 1990; 66: 554–564.
- MacLennan DH, Brandl CJ, Korczak B, Green NM. Amino-acid sequence of a Ca2+ + Mg2+-dependent ATPase from rabbit muscle sarcoplasmic reticulum, deduced from its complementary DNA sequence. Nature 1985; 316: 696–700.
- Gorski PA, Ceholski DK, Hajjar RJ. Altered myocardial calcium cycling and energetics in heart failure – a rational approach for disease treatment. Cell Metab 2015; 21: 183–194.
- MacLennan DH. Purification and properties of an adenosine triphosphatase from sarcoplasmic reticulum. J Biol Chem 1970; 245: 4508–4518.
- Katz AM. Cardiomyopathy of overload. A major determinant of prognosis in congestive heart failure. N Engl J Med 1990; 322: 100–110.
- Braunwald E. Shattuck lecture – cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med 1997; 337: 1360–1369.
- Roger VL. Epidemiology of heart failure. Circ Res 2013; 113: 646–659.
- Baker DL, Hashimoto K, Grupp IL, Ji Y, Reed T, Loukianov E, et al. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ Res 1998; 83: 1205–1214.
- del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999; 100: 2308–2311.
- Studeli R, Jung S, Mohacsi P, Perruchoud S, Castiglioni P, Wenaweser P, et al. Diastolic dysfunction in human cardiac allografts is related with reduced SERCA2a gene expression. Am J Transplant 2006; 6: 775–782.
- Barkley GL, Moran JE, Takanashi Y, Tepley N. Techniques for DC magnetoencephalography. J Clin Neurophysiol 1991; 8: 189–199.
- Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol 2008; 51: 1112–1119.
- Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011; 124: 304–313.
- Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016; 387: 1178–1186.
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004; 110: 705–712.
- Mao W, Fukuoka S, Iwai C, Liu J, Sharma VK, Sheu SS, et al. Cardiomyocyte apoptosis in autoimmune cardiomyopathy: mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am J Physiol Heart Circ Physiol 2007; 293: H1636–H1645.
- Li SY, Gilbert SA, Li Q, Ren J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J Mol Cell Cardiol 2009; 47: 247–255.
- Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol 2007; 50: 1029–1037.
- Force T, Kerkela R. Cardiotoxicity of the new cancer therapeutics – mechanisms of, and approaches to, the problem. Drug Discov Today 2008; 13: 778–784.
- Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 2007; 116: 1226–1233.
- Ortega A, Rosello-Lleti E, Tarazon E, Molina-Navarro MM, Martinez-Dolz L, Gonzalez-Juanatey JR, et al. Endoplasmic reticulum stress induces different molecular structural alterations in human dilated and ischemic cardiomyopathy. PLoS One 2014; 9: e107635.
- Dally S, Monceau V, Corvazier E, Bredoux R, Raies A, Bobe R, et al. Compartmentalized expression of three novel sarco/endoplasmic reticulum Ca2+ATPase 3 isoforms including the switch to ER stress, SERCA3f, in non-failing and failing human heart. Cell Calcium 2009; 45: 144–154.
- Duan Q, Ni L, Wang P, Chen C, Yang L, Ma B, et al. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell 2016; 15: 625–633.
- Duan Q, Yang L, Gong W, Chaugai S, Wang F, Chen C, et al. MicroRNA-214 is upregulated in heart failure patients and suppresses XBP1-mediated endothelial cells angiogenesis. J Cell Physiol 2015; 230: 1964–1973.
- Lu Z, Xu X, Fassett J, Kwak D, Liu X, Hu X, et al. Loss of the eukaryotic initiation factor 2alpha kinase general control nonderepressible 2 protects mice from pressure overload-induced congestive heart failure without affecting ventricular hypertrophy. Hypertension 2014; 63: 128–135.
- Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, et al. A thrombospondin-dependent pathway for a protective ER stress response. Cell 2012; 149: 1257–1268.
- Ibler KS, Jemec GB. Cumulative life course impairment in other chronic or recurrent dermatologic diseases. Curr Probl Dermatol 2013; 44: 130–136.
- Langley RG, Feldman SR, Han C, Schenkel B, Szapary P, Hsu MC, et al. Ustekinumab significantly improves symptoms of anxiety, depression, and skin-related quality of life in patients with moderate-to-severe psoriasis: results from a randomized, double-blind, placebo-controlled phase III trial. J Am Acad Dermatol 2010; 63: 457–465.
- Yazici K, Baz K, Yazici AE, Kokturk A, Tot S, Demirseren D, et al. Disease-specific quality of life is associated with anxiety and depression in patients with acne. J Eur Acad Dermatol Venereol 2004; 18: 435–439.
- Gordon-Smith K, Green E, Grozeva D, Tavadia S, Craddock N, Jones L. Genotype-phenotype correlations in Darier disease: a focus on the neuropsychiatric phenotype. Am J Med Genet B Neuropsychiatr Genet 2018; 177: 717–726.
- Earls LR, Bayazitov IT, Fricke RG, Berry RB, Illingworth E, Mittleman G, et al. Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J Neurosci 2010; 30: 15843–15855.
- Jacobsen NJ, Lyons I, Hoogendoorn B, Burge S, Kwok PY, O’Donovan MC, et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet 1999; 8: 1631–1636.
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 2008; 31: 454–463.
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 2002; 3: 862–872.
- Dahl R. A new target for Parkinson’s disease: Small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg Med Chem 2017; 25: 53–57.
- Brini M, Carafoli E, Cali T. The plasma membrane calcium pumps: focus on the role in (neuro)pathology. Biochem Biophys Res Commun 2017; 483: 1116–1124.
- Baba-Aissa F, Raeymaekers L, Wuytack F, De Greef C, Missiaen L, Casteels R. Distribution of the organellar Ca2+ transport ATPase SERCA2 isoforms in the cat brain. Brain Res 1996; 743: 141–153.
- Campbell AM, Wuytack F, Fambrough DM. Differential distribution of the alternative forms of the sarcoplasmic/endoplasmic reticulum Ca(2+)-ATPase, SERCA2b and SERCA2a, in the avian brain. Brain Res 1993; 605: 67–76.
- Salvador JM, Berengena M, Sepulveda MR, Mata AM. Distribution of the intracellular Ca(2+)-ATPase isoform 2b in pig brain subcellular fractions and cross-reaction with a monoclonal antibody raised against the enzyme isoform. J Biochem 2001; 129: 621–626.
- Craddock N, Owen M, Burge S, Kurian B, Thomas P, McGuffin P. Familial cosegregation of major affective disorder and Darier’s disease (keratosis follicularis). Br J Psychiatry 1994; 164: 355–358.
- Cheour M, Zribi H, Abdelhak S, Drira S, Ben Osman A. Les manifestations neuropsychiatriques de la maladie de Darier: résultat préliminaire d’une étude epidémioclinique et génétique de huit familles. Encephale 2009; 35: 32–35.
- Wang SL, Yang SF, Chen CC, Tsai PT, Chai CY. Darier’s disease associated with bipolar affective disorder: a case report. Kaohsiung J Med Sci 2002; 18: 622–626.
- Jones I, Jacobsen N, Green EK, Elvidge GP, Owen MJ, Craddock N. Evidence for familial cosegregation of major affective disorder and genetic markers flanking the gene for Darier’s disease. Mol Psychiatry 2002; 7: 424–427.
- Cederlof M, Bergen SE, Langstrom N, Larsson H, Boman M, Craddock N, et al. The association between Darier disease, bipolar disorder, and schizophrenia revisited: a population-based family study. Bipolar Disord 2015; 17: 340–344.
- Dodiuk-Gad RP, Cohen-Barak E, Khayat M, Milo H, Amariglio-Diskin L, Danial-Faran N, et al. Darier disease in Israel: combined evaluation of genetic and neuropsychiatric aspects. Br J Dermatol 2016; 174: 562–568.
- Berridge MJ, Bootman MD, Lipp P. Calcium – a life and death signal. Nature 1998; 395: 645–648.
- Atack JR, Broughton HB, Pollack SJ. Inositol monophosphatase – a putative target for Li+ in the treatment of bipolar disorder. Trends Neurosci 1995; 18: 343–349.
- Jope RS, Song L, Li PP, Young LT, Kish SJ, Pacheco MA, et al. The phosphoinositide signal transduction system is impaired in bipolar affective disorder brain. J Neurochem 1996; 66: 2402–2409.
- Ngo J, Haber R. Exacerbation of Darier disease by lithium carbonate. J Cutan Med Surg 2010; 14: 80–84.
- Rubin MB. Lithium-induced Darier’s disease. J Am Acad Dermatol 1995; 32: 674–675.
- Sule N, Teszas A, Kalman E, Szigeti R, Miseta A, Kellermayer R. Lithium suppresses epidermal SERCA2 and PMR1 levels in the rat. Pathol Oncol Res 2006; 12: 234–236.
- Liu LH, Boivin GP, Prasad V, Periasamy M, Shull GE. Squamous cell tumors in mice heterozygous for a null allele of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump. J Biol Chem 2001; 276: 26737–26740.
- Endo Y, Uzawa K, Mochida Y, Shiiba M, Bukawa H, Yokoe H, et al. Sarcoendoplasmic reticulum Ca(2+) ATPase type 2 downregulated in human oral squamous cell carcinoma. Int J Cancer 2004; 110: 225–231.
- Pacifico F, Ulianich L, De Micheli S, Treglia S, Leonardi A, Vito P, et al. The expression of the sarco/endoplasmic reticulum Ca2+-ATPases in thyroid and its down-regulation following neoplastic transformation. J Mol Endocrinol 2003; 30: 399–409.
- Gelebart P, Kovacs T, Brouland JP, van Gorp R, Grossmann J, Rivard N, et al. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J Biol Chem 2002; 277: 26310–26320.
- Korosec B, Glavac D, Rott T, Ravnik-Glavac M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet Cytogenet 2006; 171: 105–111.
- Prasad V, Boivin GP, Miller ML, Liu LH, Erwin CR, Warner BW, et al. Haploinsufficiency of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump, predisposes mice to squamous cell tumors via a novel mode of cancer susceptibility. Cancer Res 2005; 65: 8655–8661.
- Li L, Tucker RW, Hennings H, Yuspa SH. Inhibitors of the intracellular Ca(2+)-ATPase in cultured mouse keratinocytes reveal components of terminal differentiation that are regulated by distinct intracellular Ca2+ compartments. Cell Growth Differ 1995; 6: 1171–1184.
- Fan L, Li A, Li W, Cai P, Yang B, Zhang M, et al. Novel role of sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed Pharmacother 2014; 68: 1141–1148.
- Wang L, Li W, Yang Y, Hu Y, Gu Y, Shu Y, et al. High expression of sarcoplasmic/endoplasmic reticulum Ca(2+)-ATPase 2b blocks cell differentiation in human liposarcoma cells. Life Sci 2014; 99: 37–43.
- Hu Z, Bonifas JM, Beech J, Bench G, Shigihara T, Ogawa H, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet 2000; 24: 61–65.
- Korman AM, Milani-Nejad N. Darier disease. JAMA Dermatol 2020 Aug 12. [Online ahead of print].
- Burge SM, Buxton PK. Topical isotretinoin in Darier’s disease. Br J Dermatol 1995; 133: 924–928.
- Burkhart CG, Burkhart CN. Tazarotene gel for Darier’s disease. J Am Acad Dermatol 1998; 38: 1001–1002.
- Abe M, Inoue C, Yokoyama Y, Ishikawa O. Successful treatment of Darier’s disease with adapalene gel. Pediatr Dermatol 2011; 28: 197–198.
- Shalita A. The integral role of topical and oral retinoids in the early treatment of acne. J Eur Acad Dermatol Venereol 2001; 15: 43–49.
- Chu S, Michelle L, Ekelem C, Sung CT, Rojek N, Mesinkovska NA. Oral isotretinoin for the treatment of dermatologic conditions other than acne: a systematic review and discussion of future directions. Arch Dermatol Res 2020 Nov 5. [E-pub ahead of print].
- Czernielewski J, Michel S, Bouclier M, Baker M, Hensby JC. Adapalene biochemistry and the evolution of a new topical retinoid for treatment of acne. J Eur Acad Dermatol Venereol 2001; 15: 5–12.
- Demczuk M, Huang H, White C, Kipp JL. Retinoic acid regulates calcium signaling to promote mouse ovarian granulosa cell proliferation. Biol Reprod 2016; 95: 70.
- Launay S, Gianni M, Diomede L, Machesky LM, Enouf J, Papp B. Enhancement of ATRA-induced cell differentiation by inhibition of calcium accumulation into the endoplasmic reticulum: cross-talk between RAR alpha and calcium-dependent signaling. Blood 2003; 101: 3220–3228.
- Wolf JE, Jr. Potential anti-inflammatory effects of topical retinoids and retinoid analogues. Adv Ther 2002; 19: 109–118.
- Letule V, Herzinger T, Ruzicka T, Molin S. Treatment of Darier disease with oral alitretinoin. Clin Exp Dermatol 2013; 38: 523–525.
- Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol 1999; 135: 423–427.
- van Dooren-Greebe RJ, van de Kerkhof PC, Happle R. Acitretin monotherapy in Darier’s disease. Br J Dermatol 1989; 121: 375–379.
- Gilgor RS, Chiaramonti A, Goldsmith LA, Lazarus GS. Evaluation of 13-cis retinoic acid in lamellar ichthyosis, pityriasis rubra pilaris and Darier’s disease. Cutis 1980; 25: 380–381, 385.
- Zavattaro E, Celasco M, Delrosso G, Ferri S, Bornacina C, Valente G, et al. Acitretin-induced acral hemorrhagic lesions in Darier-White disease. Cutis 2014; 94: E1–E5.
- Larbre B, Nicolas JF, Frappaz A, Thivolet J. Cyclosporine et maladie de Darier. Ann Dermatol Venereol 1993; 120: 310–311.
- Gruber SJ, Cornea RL, Li J, Peterson KC, Schaaf TM, Gillispie GD, et al. Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J Biomol Screen 2014; 19: 215–222.
- Dong Z, Saikumar P, Weinberg JM, Venkatachalam MA. Calcium in cell injury and death. Annu Rev Pathol 2006; 1: 405–434.