Dyskeratosis congenita (DC) is a telomere biology disorder characterized by a diagnostic triad of abnormal skin pigmentation, dysplastic nails, and oral leukoplakia (1). Moreover, patients with DC are at an increased risk of developing multisystemic disease, including bone marrow failure (BMF), myelodysplastic syndrome (MDS), acute myeloid leukaemia (AML), solid tumours (most often squamous cell carcinoma of the head and neck), and pulmonary fibrosis (1). These clinical features are thought to arise owing to replicative senescence due to severely shortened telomeres. Loss of function in cells with high turnover, such as dermal and ectodermal stem cells, may explain the mucocutaneous manifestations (2).
Overall, 16 different genes encoding critical components of the telomere have been found to be causative in DC (DKC1, TERC, TERT, NOP10, NHP2, ACD, TINF2, POT1, CTC1, STN1, NAF1, WRAP53, PARN, TPP1, RAP1 and RTEL1) (1).
We report here a patient with pulmonary fibrosis, atypical dermatological findings, and a novel pathogenic variant in the RTEL1 gene.
CASE REPORT
A 49-year-old woman with a history of deep vein thrombosis, chronic kidney failure, multiple contact allergies, atopic dermatitis (AD), and pulmonary fibrosis was referred to our department with severe AD. Since age < 2 years the patient had AD with intermittent flare-ups. During the first 2 years of referral the patient was treated with methotrexate, azathioprine and mycophenolate mofetil, but all had to be discontinued due to herpes simplex virus infection or gastrointestinal and respiratory side-effects. Furthermore, treatment with ultraviolet B (UVB), psoralen plus UVA (PUVA), and prednisolone were insufficient to control the disease. Over the past year the patient’s eczema cleared due to dupilumab treatment.
The patient was diagnosed with pulmonary fibrosis (PF) in 2015 (Fig. 1). A provisional diagnosis of fibrotic allergic alveolitis was made, based on exposure to birds, relative lymphocytosis in bronchoalveolar lavage and unspecific interstitial inflammation and fibrosis. Due to a decline in lung function, no family history of PF and no history of smoking she was referred to genetic counselling in 2020. Her family history was unremarkable, and there had been no reports of DC-related disease or cancers among family members for 3 generations. The genetic test result revealed that the patient carried a germline variant in the RTEL1 gene (c.2299C>T, p.Arg767*). The variant has not been reported previously in the literature. To confirm the diagnosis of DC a telomere length analysis using flow cytometry and fluorescent in situ hybridization (flow-FISH) was performed. Indeed, the results showed markedly shortened telomeres in all cell lines. Subsequently, segregation analysis revealed that the variant was inherited from the patient’s mother (age 72 years), who is healthy, and that a number of healthy relatives were carriers.
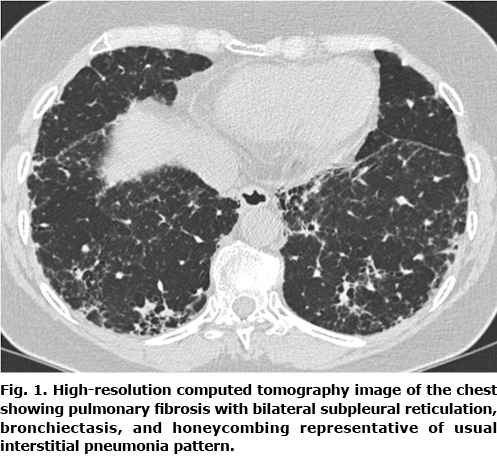
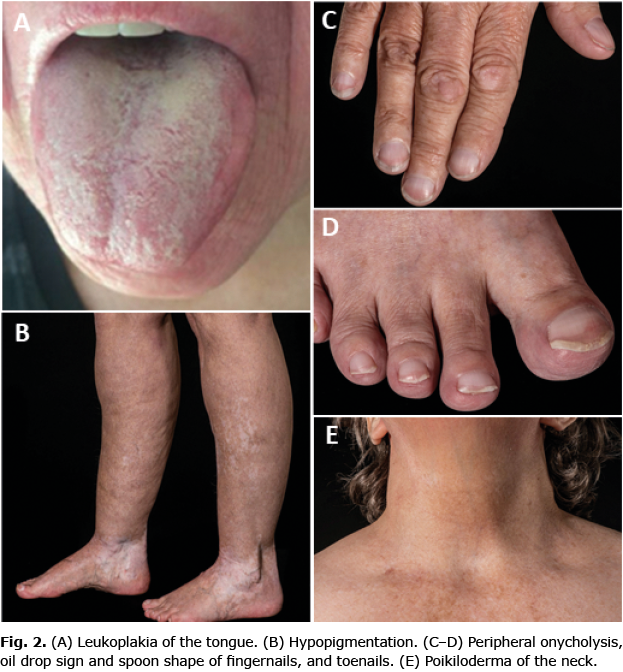
In concordance with DC, the current patient has leukoplakia of the tongue, onycholysis, poikiloderma of the neck, abnormal pigmentation, short stature (152 cm), grey hair, alopecia, affected tear ducts, and PF (see dermatological features in Fig. 2). Patients with pulmonary fibrosis due to DC experience exertional dyspnoea and cough with restrictive lung function test and reduced diffusion capacity, which correlates with the current patient’s symptoms and clinical findings (3).
DISCUSSION
DC was first described as a genodermatosis in 1906 by Zinsser (4). The case report included only men with the classic triad (4). The first female case was described 57 years later (5). This may reflect the inheritance of DC, as the disease is often X-linked recessive, and males are up to 13 times more likely to be affected (6). Other forms of inheritance include autosomal dominant and autosomal recessive, which are true for mutations in the RTEL1 gene (7). Moreover, homozygous mutations in the RTEL1 gene tend to be associated with Hoyeraal-Hreidarsson syndrome, a phenotypically severe variant of DC with very short telomeres, while heterozygous mutations are more frequently identified in patients with familial PF (3). Apart from the classic triad of DC, other cutaneous manifestations include premature hair greying, loss of scalp hair or eyebrows, palmoplantar hyperkeratosis with absent papillary relief, skin cancers including basal cell carcinoma or squamous cell carcinoma, white striae, and hyperhidrosis (8).
As denoted previously, AD has not previously been described in patients with DC. Interestingly, in a study of telomerase activity in patients with atopic dermatitis, the study group found increased telomerase activity and shortened telomere length in T cells (9). As patients with DC already have shortened telomeres, this finding could explain how AD may be accelerated in patients with DC.
Due to the rarity of the disease, genetic heterogeneity, and variation in phenotypic penetrance, the correct diagnosis may often be delayed, as was the case for the current patient. However, correct and early detection of the disease is crucial due to the potentially lethal prognosis. Close multidisciplinary and long-term follow-ups are essential to detect and possibly treat new manifestations (1).
With this rare case of DC with a novel pathogenic variant in the RTEL1 gene, further study is required into the dermatological and pulmonary signs of DC that may lead to proper genetic testing and diagnosis.
The authors have no conflicts of interest to declare.
REFERENCES
- Savage SA. Beginning at the ends: telomeres and human disease. F1000Res 2018; 7: 524.
- Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol 2016; 13: 696–706.
- Wang P, Xu Z. Pulmonary fibrosis in dyskeratosis congenita: a case report with PRISMA-compliant systematic review. BMC Pulm Med 2021; 21: 279.
- Zinsser F. Atropha cutis reticularis cum pigmentatione, dystrophia ungiumet leukoplakia oris. Ikongr Dermatol 1906; 5: 219–223.
- Sorrow JM Jr, Hitch JM. Dyskeratosis congenita. First report of its occurrence in a female and a review of the literature. Arch Dermatol 1963; 13: 696–706.
- Kumar S, Suthar R. Dyskeratosis congenita. JK Science 2013; 15: 56–58.
- Vannier JB, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends Cell Biol 2014; 24: 416–425.
- AlSabbagh MM. Dyskeratosis congenita: a literature review. J Dtsch Dermatol Ges 2020; 18: 943–967.
- Wu K, Higashi N, Hansen ER, Lund M, Bang K, Thestrup-Pedersen K. Telomerase activity is increased and telomere length shortened in T cells from blood of patients with atopic dermatitis and psoriasis. J Immunol 2000; 165: 4742–4747.