RESEARCH ARTICLE
Advanced glycation end products induce inflammaging in periodontal ligament fibroblasts through RAGE/AKT/mTOR/glycolysis pathway
Lin Xionga, Jiayu Shub, Hongli Gaoa, Yufeng Qina, Yuehan Zhanga, Xuelian Changa, Qiang Dongb and Helin Chenb
aCollege of Stomatology of Guizhou Medical University, Guiyang, China; bDepartment of Prosthodontics, Stomatological Hospital of Guizhou Medical University, Guiyang, China
ABSTRACT
Background: Inflammaging plays a pivotal role in the pathogenesis of multiple age-related diseases, including periodontitis. Advanced glycation end products (AGEs) are known to induce inflammaging and exacerbate periodontitis. However, the mechanisms by which AGEs promote inflammaging remain unclear. This study aimed to investigate the mechanisms underlying AGE-induced inflammaging.
Methods and results: Human periodontal ligament fibroblasts (hPDLFs) were extracted and stimulated with lipopolysaccharide (LPS), with prior treatment using AGEs. The expression of pro-inflammatory cytokines was measured to explore the role of AGEs in LPS-induced inflammation. Subsequently, hPDLFs were treated with AGEs and pre-incubated with 2-deoxyglucose (2-DG, a glycolysis inhibitor), Ly294002 (an AKT/mTOR pathway inhibitor), and FPS-ZM1 (a receptor for advanced glycation end product [RAGE] antagonist) to assess the levels of inflammaging markers, glycolysis, AKT/mTOR pathway activation, and RAGE expression, along with the potential relationships among these factors. Our findings demonstrated that AGEs significantly increased the expression of pro-inflammatory cytokines in response to LPS stimulation. Additionally, AGEs alone elevated the levels of inflammaging factors, including cell senescence, senescence-associated secretory phenotype factors, SA-β-Gal expression, glycolysis markers, and AKT/mTOR pathway activation. Furthermore, inhibiting glycolysis reduced AGE-induced inflammaging, while blocking the AKT/mTOR pathway, suppressed both AGE-induced inflammaging and glycolysis. Antagonizing RAGE effectively blocked AGE-induced inflammaging, glycolysis, and AKT/mTOR pathway activation.
Conclusions: Our study indicated that AGE-induced inflammaging through binding to RAGE to activate the AKT/mTOR pathway and eventually enhancing glycolysis level, which may contribute to the increased inflammatory response triggered by LPS. These findings suggest that inflammaging is a critical mechanism through which AGEs exacerbate periodontitis.
KEYWORDS: periodontitis; advanced glycation end products; glycolysis; inflammaging; Akt/mTOR pathway
Citation: ACTA ODONTOLOGICA SCANDINAVICA 2025; VOL. 84: 479–490. DOI: https://doi.org/10.2340/aos.v84.44581.
Copyright: © 2025 The Author(s). Published by MJS Publishing on behalf of Acta Odontologica Scandinavica Society. This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), allowing third parties to copy and redistribute the material in any medium or format and to remix, transform, and build upon the material, with the condition of proper attribution to the original work.
Received: 21 April 2025; Accepted: 27 July 2025; Published: 21 August 2025.
CONTACT: Helin Chen helinchen0509@163.com College of Stomatology of Guizhou Medical University, Guiyang, 550004, Guizhou, China.
Competing interests and funding: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This study was supported by the National Natural Science Foundation of China (82160186), the Science and Technology Program of Guizhou Province (Guizhou Science and Technology Cooperation Foundation-ZK[2021]General 433).
Introduction
Periodontitis is a chronic infectious disease affecting periodontal tissues [1], with a global prevalence rate of approximately 45–50%, and this figure continues to rise [2]. Currently, periodontitis ranks as one of the most prevalent diseases worldwide and is a leading cause of tooth loss in older adults [3]. Numerous systemic diseases, such as diabetes, can aggravate periodontitis. There is emerging evidence to support the existence of a two-way relationship between diabetes and periodontitis, with diabetes increasing the risk for periodontitis, and periodontal inflammation negatively affecting glycaemic control [4]. Diabetes mellitus exacerbates periodontitis, with advanced glycation end products (AGEs) identified as a significant contributing factor [5, 6]. AGEs, a heterogeneous group of compounds, interact with the receptor for advanced glycation end products (RAGE) to modulate cellular metabolism [7, 8]. However, the precise mechanisms by which AGEs exacerbate periodontitis remain incompletely understood.
Periodontitis is an age-related disease, in which cellular senescence plays a significant role in its progression [9]. Inflammaging, a chronic state of low-grade inflammation, is a distinct form of senescence that affects the aging process and contributes to various age-related diseases [10]. Cellular senescence and the senescence-associated secretory phenotype (SASP) are key features of inflammaging [11]. Under certain stimuli, the low-grade inflammatory state can escalate into a more severe inflammatory response, resulting in increased tissue damage. Consequently, inflammaging is implicated in the pathogenesis of numerous inflammatory diseases, including osteoarthritis, rheumatoid arthritis, and periodontitis [3, 12, 13]. Studies have shown that inflammaging is linked to periodontal epithelial barrier dysfunction [14]. Knockout of TLR9 to suppress inflammaging has been found to mitigate alveolar bone resorption [15]. Recent research suggests that AGEs may induce inflammaging [16]. Thus, we hypothesize that AGEs exacerbate periodontitis by inducing inflammaging.
Mounting evidence underscores the role of glycolysis in cellular senescence [17, 18]. Glycolysis is a critical metabolic pathway for cellular energy production. Upon glucose uptake, the PI3K/Akt/mTOR signaling pathway is activated, which upregulates the expression of glucokinase (GK), initiating glucose catabolism [19]. Several factors, including AGEs [20], can redirect cellular metabolism toward glycolysis. Elevated glycolysis has been shown to contribute to inflammatory dysfunction, SASP secretion, and cellular senescence [21, 22]. However, whether glycolysis is involved in AGE-induced inflammaging remains unclear.
In this study, human periodontal ligament fibroblasts (hPDLFs) were isolated and stimulated with AGEs to investigate AGE-induced inflammaging by assessing cellular senescence and SASP secretion. Additionally, the roles of the PI3K/Akt pathway and glycolysis in AGE-induced inflammaging were examined.
Materials and methods
AGE-BSA preparation
Bovine serum albumin (BSA, Solebao, Beijing, China) at 50 mg/mL and D-glyceraldehyde (0.1 mol/L, Aladding, Shanghai, China) were dissolved in sterile phosphate buffer (0.2 mol/L, pH 7.4). The mixture was filtered through a 0.22-µm filter membrane and sterilized and then incubated in the dark at 37°C for 7 days. Following incubation, the mixture was dialyzed using phosphate-buffered saline (PBS, pH 7.4) to remove free glyceraldehyde and unbound low molecular weight reactants. As a control, 50 mg/mL BSA without glyceraldehyde was incubated under the same conditions. Given the fluorescent nature of AGEs, the fluorescence intensity of the AGE-BSA solution was measured using a fluorescence spectrophotometer to differentiate between the AGE-BSA and the control solution (excitation wavelength 370 nm, emission wavelength 440 nm, and slit width 3 nm). The fluorescence intensity of the AGE-BSA solution was found to be 45 times higher than that of the control.
Cell culture and identification
Twenty healthy premolars extracted for orthodontic treatment were collected and stored in a solution containing 100 U/mL penicillin and 100 mg/mL streptomycin. The periodontal ligament tissue was carefully scraped and cut into small pieces. The samples were then digested in type I collagenase (Invitrogen) for 30 minutes, followed by incubation in dulbecco’s modified eagle medium, DMEM supplemented with 15% fetal bovine serum (FBS), 1% 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C with 5% CO2. Cells were passaged using 0.25% trypsin-ethylene diamine tetraacetic acid, EDTA (Gibco, USA). The third to sixth generations of cells were used in this study. CD105, CD90, CD31, and CD14 were selected for fibroblast cell identification. Patients enrolled in the study were aged 18–25 years and provided signed informed consent. The study was approved by the Ethics Committee of the School of Stomatology at Guizhou Medical University, Ethical Review Document No. 31 of 2021
Quantitative real‐time polymerase chain reaction
Trizol (Thermo Fisher, USA) was used to extract total RNA from hPDLFs. The obtained RNA was then reverse transcribed into cDNA following the protocol of the PrimeScript RT Reagent Kit (Takara, Japan). Finally, quantitative real‐time polymerase chain reaction (qRT-PCR) was performed to measure the mRNA levels using the TB Green® Premix Ex Taq™ kit. The specific primers are listed below.
| Gene | Forward sequence (5‘-3‘) | Reverse sequence (5‘-3‘) |
| IL-1β | ATC AGC ACC TCT CAA GCA G | AGT CCA CAT TCA GCA CAG G |
| IL-6 | CAA TAA CCA CCC CTG ACC | GCG CAG AAT GAG ATG AGT |
| TNF-α | GGA AAG GAC ACC ATG AGC | CCA CGA TCA GGA AGG AGA |
| β-actin | CCT GGC ACC CAG CAC AAT | GGG CCG GAC TCG TCA TAC |
Western blotting
Proteins were extracted from hPDLFs using a total protein extraction kit (Solarbio, Beijing, China) according to the manufacturer’s protocol. Protein samples from each group were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to a 0.45 µm polyvinylidene difluoride, PVDF membrane (Millipore) via electroblotting. The membranes were blocked with 5% milk for 1 hour, followed by overnight incubation at 4°C in the dark with primary antibodies against β-actin (1:3,000; Wuhan Sanying), HKII (1:2,000, Abcam, USA), PKM2 (1:2,000, Abcam, USA), GLUT1 (1:1,000, Abcam, USA), LDHA (1:2,000, Abcam, USA), P16 (1:2,000; Wuhan Sanying), P21 (1:2,000; Wuhan Sanying), P53 (1:2,000; Wuhan Sanying), RAGE (1:2,000; Wuhan Sanying), AKT2 (1:2,000; Wuhan Sanying), p-AKT (1:2,000; Wuhan Sanying), mTOR (1:2,000; Wuhan Sanying), and p-mTOR (1:2,000; Wuhan Sanying). The membranes were then incubated with a secondary antibody (1:5,000, Wuhan Sanying) conjugated to horseradish peroxidase (HRP) at room temperature for 1 hour. The bound antibody was detected using the West Pico Chemiluminescent Substrate System (SuperSignal, BioSpectrum®310 Imaging System, USA). The resulting images were analyzed with ImageJ software (National Institutes of Health).
Measurement of glucose uptake and lactic acid production
The measurement of glucose and lactic acid content in the supernatant was performed according to the protocols of the glucose detection kit (Nanjing Jiancheng, China) and the lactate detection kit (Nanjing Jiancheng, China). The glucose uptake was calculated as follows: glucose content (t0) – glucose content (t1). The lactic acid production was calculated as follows: lactic acid content (t1) – lactic acid content (t0).
SA-β-Gal staining
SA-β-Gal staining was performed using the SA-β-Gal staining kit (Beyotime, China). Cells were washed three times with PBS and then fixed at room temperature with 4% paraformaldehyde (PFA) for 15 minutes. Following fixation, cells were incubated overnight in the dark at 37°C in a working solution containing 0.05 mg/mL X-gal. After rinsing with PBS, the cells were observed under a microscope at 100x magnification. Five random fields from each sample were selected and analyzed using ImageJ software. SA-β-Gal positive cells were stained blue.
ELISA
The secretion levels of IL-1β, IL-6, and TNF-α were determined using the respective ELISA kits (Elabscience, China). A volume of 100 μL/well of cell supernatant was added to each well of a 96-well plate and incubated at 37°C for 90 minutes. Following incubation, 100 μL/well of biotinylated detection antibody was added to each well and incubated for 60 minutes. The plate was washed five times, and 100 μL/well of HRP conjugate was added to each well and incubated for 30 minutes. After five additional washes, substrate reagent was added and incubated for 15 minutes, followed by the addition of stop solution. Finally, the optical density (OD) of each well was measured at a wavelength of 450 nm using an enzyme marker.
Statistical analysis
All tests were conducted in triplicate, and the data are presented as the mean ± standard error of the mean. One-way ANOVA was used to compare differences between groups, with p < 0.05 considered statistically significant. All statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS) software (version 20 for Windows, SPSS Inc., USA).
Results
AGE-induced inflammaging in hPDLFs
hPDLFs were isolated and identified using flow cytometry with antibodies against CD14, CD31, CD90, and CD105, and the results are shown in Figure 1. Figure 1A and 1B displays the long and stringy morphology of hPDLFs at passages p0 and p1. Figure 1C, 1D, 1E, and 1F shows that the percentages of CD90, CD105, CD14, and CD31 positive cells are 99.0, 99.4, 0.14, and 0.051%, respectively. These findings confirm that the cells extracted from periodontal tissue are derived from mesenchymal stem cells, thus identifying them as periodontal ligament fibroblasts.
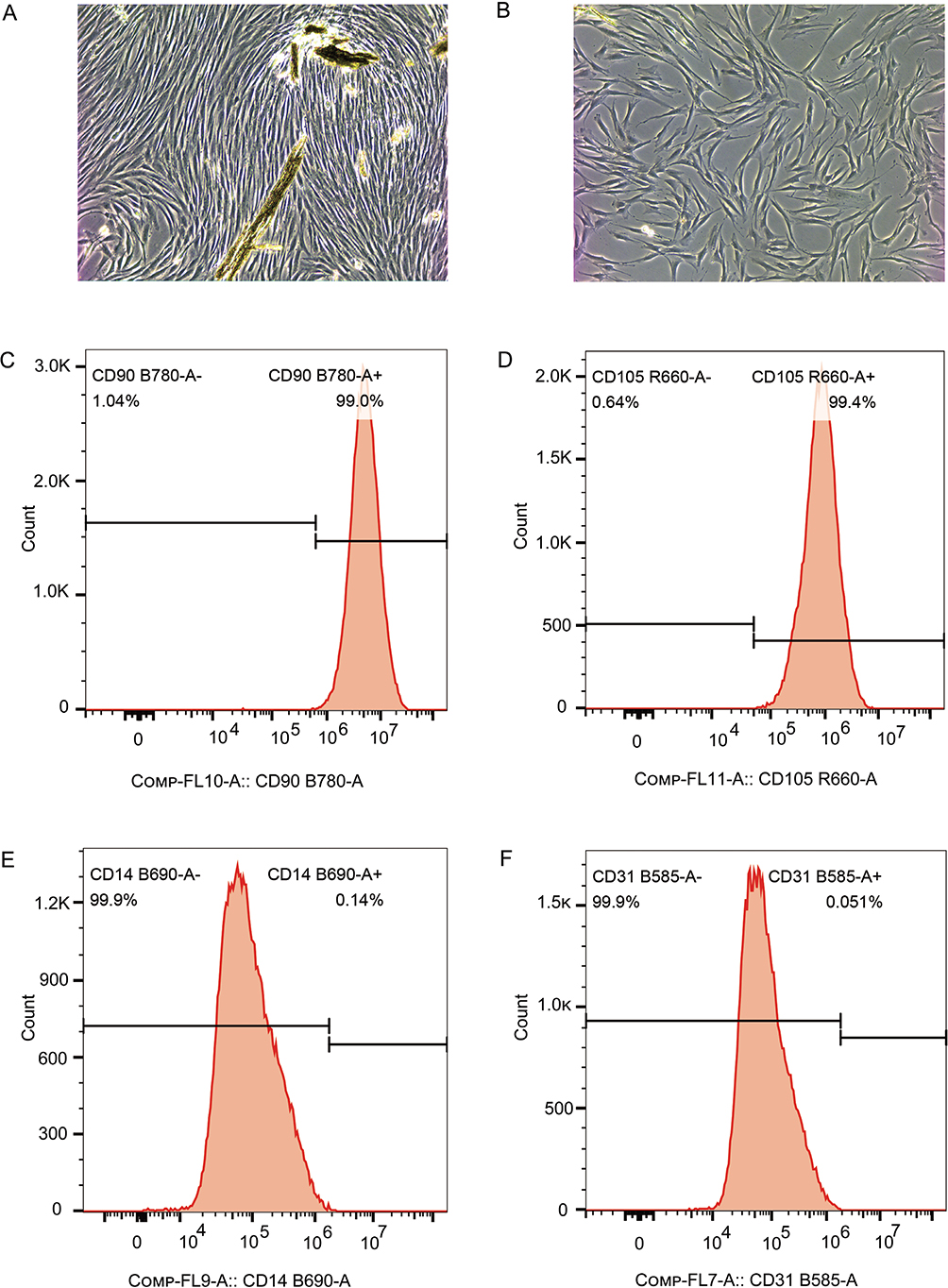
Figure 1. Extraction and identification of hPDLFs. (A) The photo of hPDLFs in generation 1 (100x). (B) The photo of hPDLFs in generation 1 (100x). (C–F) Results of the expression of surface antigens CD90, CD105, CD14, and CD31 analyzed by flow cytometry, respectively. hPDLFs: human periodontal ligament fibroblasts.
hPDLFs from p3 to p6 were stimulated with 200 µg/mL AGEs, and the expression of IL-1β, IL-6, TNF-α, p16, p21, p53, and β-galactosidase activity was measured by q-PCR, ELISA, Western blotting (WB), and SA-β-Gal staining, with the results presented in Figure 2. As shown in Figure 2, after AGE administration, the expression levels of SASP factors, including IL-1β, IL-6, TNF-α, as well as p16, p21, and p53, increased gradually, peaked at 24 hours, and then decreased in a time-dependent manner. Additionally, β-galactosidase activity was elevated, suggesting that AGEs could induce inflammaging in hPDLFs.
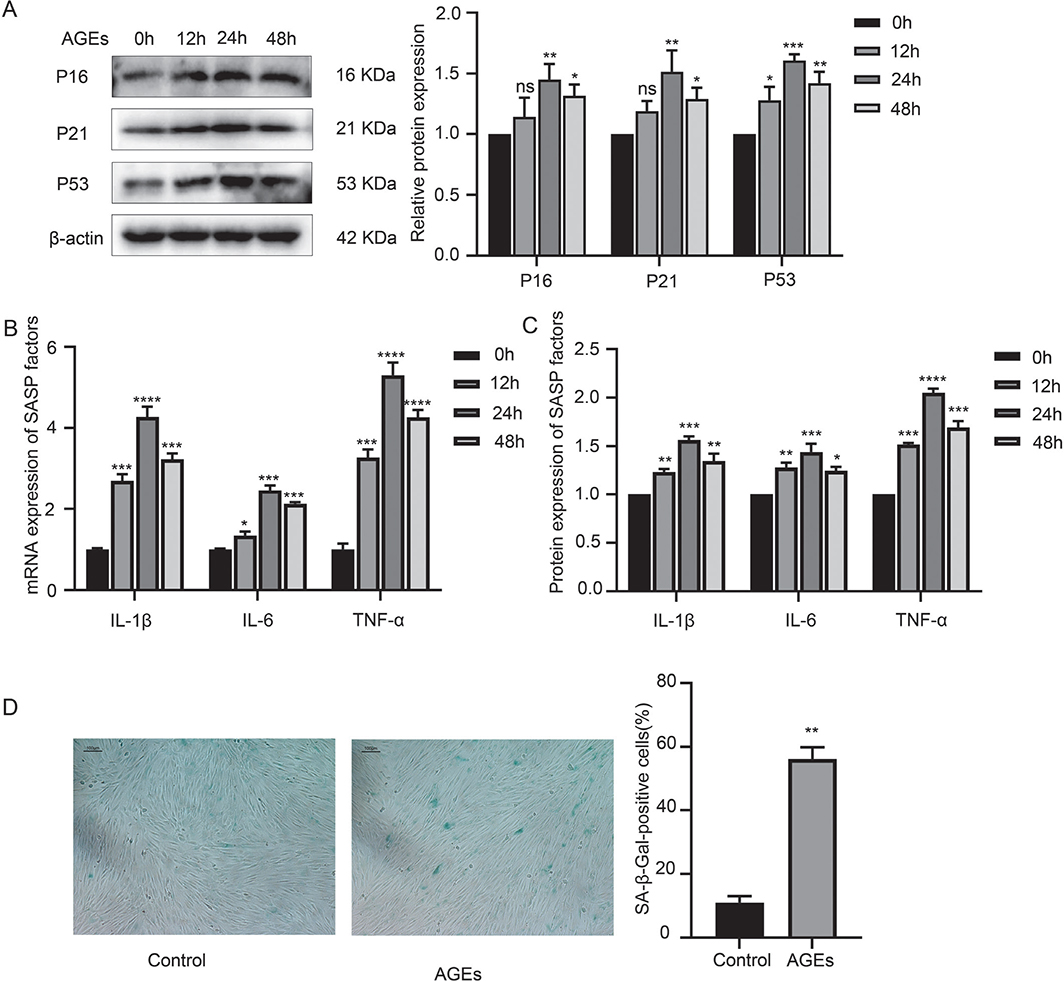
Figure 2. AGE-induced inflammaging in hPDLFs. (A) Western blotting of senescence characteristic proteins of p16, p21, and p53 in the control (oh) group and AGE group (12 h, 24 h, and 48 h). (B) mRNA expression of SASP factors, including IL-1β, IL-6, TNF-α in the control (oh) group and AGE group (12 h, 24 h, and 48 h). (C) Protein expression of SASP factors, including IL-1β, IL-6, and TNF-α in the control (oh) group and AGE group (12 h, 24 h, and 48 h). (D) SA-β-Gal staining in the control and AGE group. AGE: advanced glycation end product; SASP: senescence-associated secretory phenotype; hPDLFs: human periodontal ligament fibroblasts. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 versus control group.
Suppression of glycolysis-alleviated AGE-induced inflammaging in hPDLFs
In addition to the inflammaging-related changes in hPDLFs following AGE stimulation, the expression of glycolytic enzymes, including hexokinase II (HKII), pyruvate kinase M2 (PKM2), glucose transporter 1 (GLUT1), and lactate dehydrogenase A (LDHA), significantly increased after 24 hours of 200 µg/mL AGE stimulation. Furthermore, lactate production and glucose uptake were also significantly elevated. These findings suggest that AGEs can enhance glycolysis in hPDLFs.
To further investigate the role of glycolysis in AGE-induced inflammaging, hPDLFs were pretreated with 2-deoxyglucose (2-DG), a glycolytic inhibitor, for 2 hours before AGE stimulation for 24 hours. The results, shown in Figure 3, indicated that 2-DG treatment reduced the expression of HKII, PKM2, GLUT1, and LDHA, as well as lactate production and glucose uptake, demonstrating that 2-DG effectively inhibits glycolysis. Moreover, the expression levels of p16, p21, p53, and β-galactosidase activity were significantly decreased in hPDLFs pretreated with 2-DG and stimulated with AGEs, compared to those exposed to AGE alone. Additionally, the levels of SASP factors, including gene and protein expression, were significantly reduced after 2-DG pretreatment. These results suggest that inhibition of glycolysis can alleviate AGE-induced inflammaging in hPDLFs.
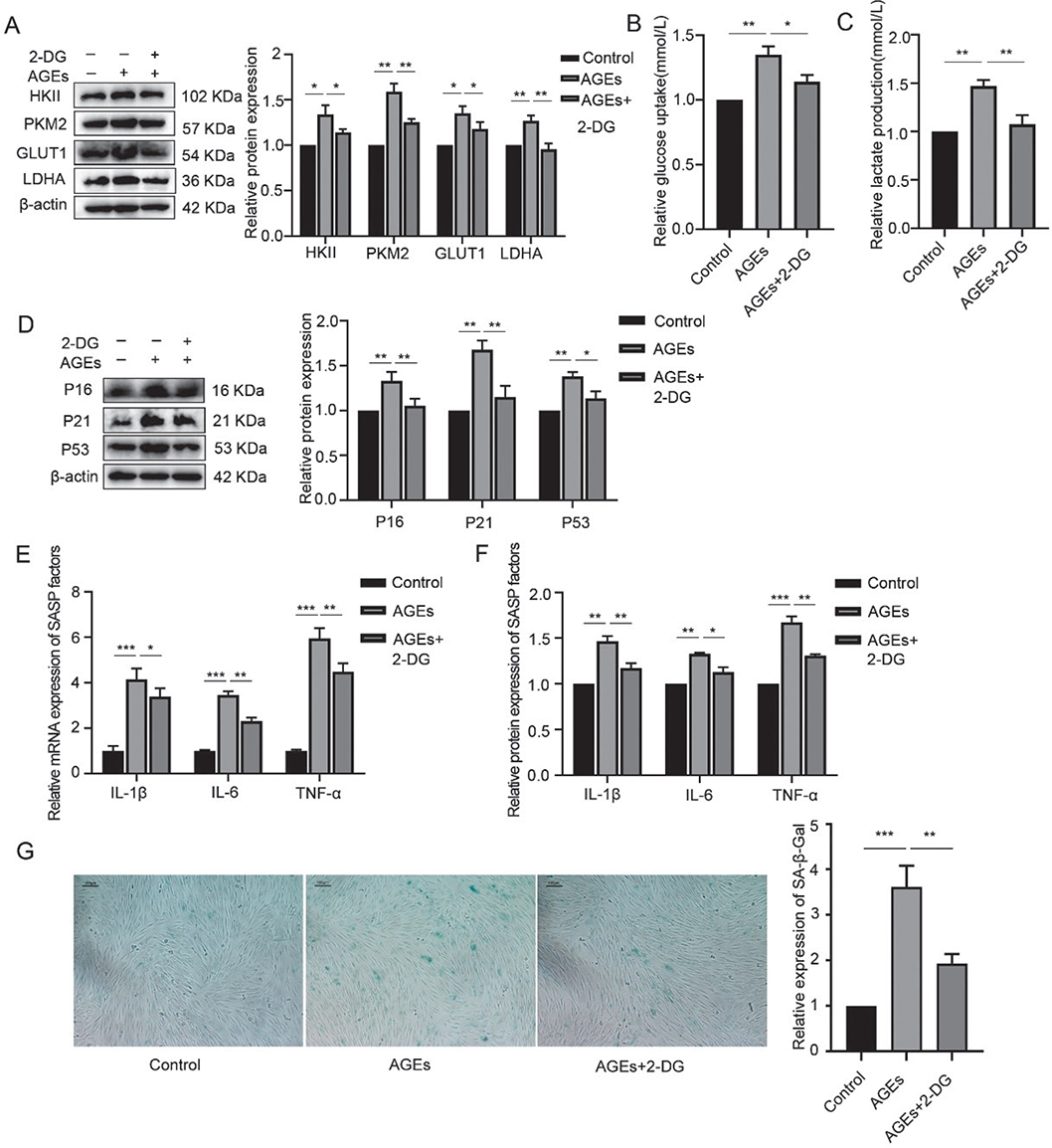
Figure 3. Suppression of glycolysis-alleviated AGE-induced inflammaging in hPDLFs. (A) Western blotting of key enzymes of glycolysis, including HKII, PKM2, GLUT1, LDHA in the control, AGEs, and AGEs+2-DG group. (B) Glucose uptake in the control, AGEs, and AGEs+2-DG group. (C) Lactate production in the control, AGEs, and AGEs+2-DG group. (D) Western blotting of senescence characteristic protein of p16, p21, and p53 in the control, AGEs, and AGEs+2-DG group. (E) mRNA expression of SASP factors including IL-1β, IL-6, and TNF-α in the control, AGEs, and AGEs+2-DG group. (F) Protein expression of SASP factors including IL-1β, IL-6, and TNF-α in the control, AGEs, and AGEs+2-DG group. (G) SA-β-Gal staining in the control, AGEs, and AGEs+2-DG group. AGEs: advanced glycation end products; SASP: senescence-associated secretory phenotype; hPDLFs: human periodontal ligament fibroblasts; 2-DG: 2-deoxyglucose. *p < 0.05, **p < 0.01, and ***p < 0.001.
Inhibition of AKT/mTOR pathway activation downregulated AGE-induced glycolysis and inflammaging
hPDLFs were treated with 200 µg/mL AGEs for 24 hours, and the activation of the AKT/mTOR signaling pathway was assessed, with the results shown in Figure 4. As illustrated in Figure 4, after AGE administration, the ratio of p-AKT/AKT and p-mTOR/mTOR significantly increased, indicating that AGEs could phosphorylate AKT and mTOR, ultimately activating the AKT/mTOR signaling pathway.
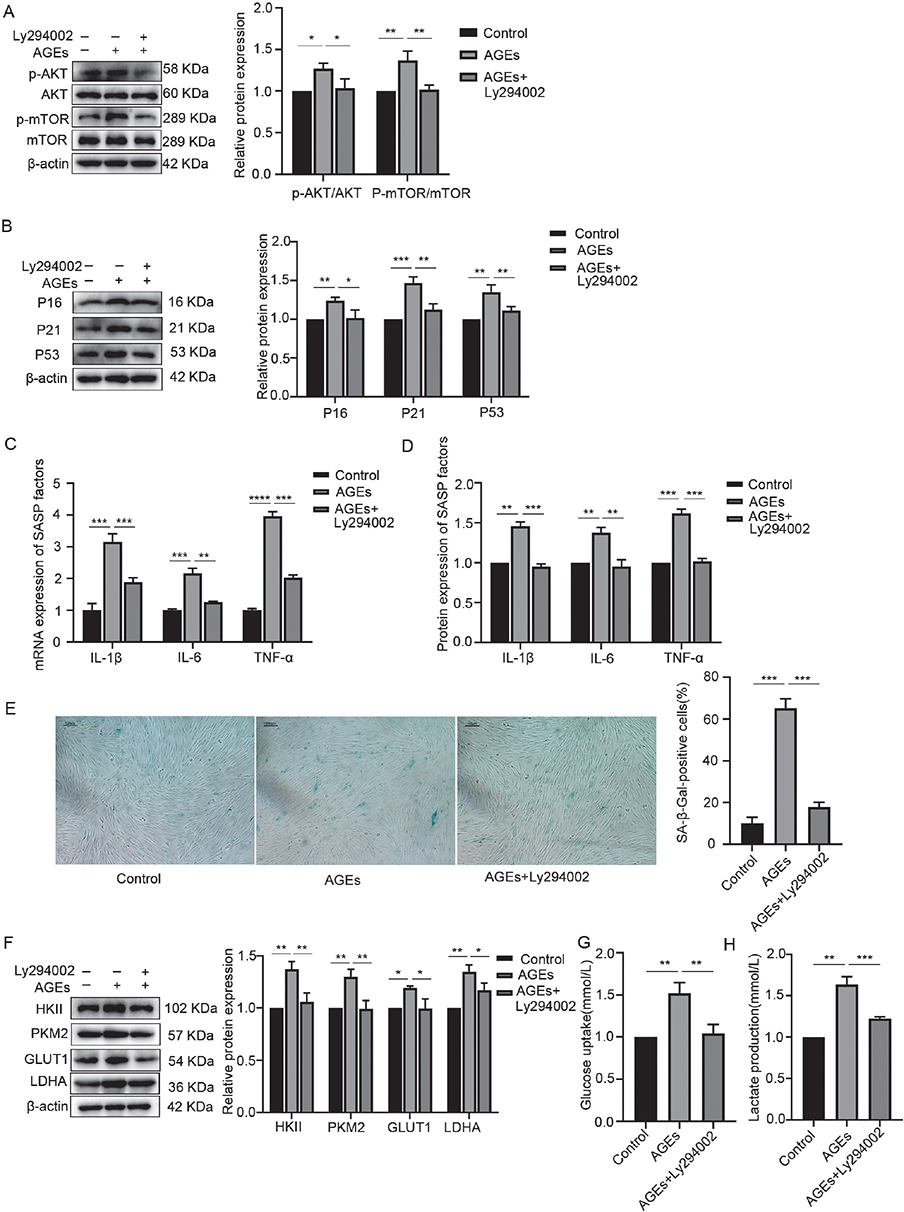
Figure 4. Inhibition of AKT/mTOR pathway activation downregulated AGE-induced glycolysis and inflammaging. (A) Western blotting of AKT, p-AKT, mTOR, p-mTOR in the control, AGEs, and AGEs+Ly294002 group. (B) Western blotting of senescence characteristic protein of p16, p21, and p53 in the control, AGEs, and AGEs+Ly294002 group. (C) mRNA expression of SASP factors including IL-1β, IL-6, and TNF-α in the control, AGEs, and AGEs+Ly294002 group. (D) Protein expression of SASP factors including IL-1β, IL-6, and TNF-α in the control, AGEs, and AGEs+Ly294002 group. (E) SA-β-Gal staining in the control, AGEs, and AGEs+Ly294002 group. (F) Relative OD value of SA-β-Gal staining in each group. (G) Western blotting of key enzymes of glycolysis including HKII, PKM2, GLUT1, and LDHA in the control, AGEs, and AGEs+Ly294002 group. (H) Glucose uptake in the control, AGEs, and AGEs+Ly294002 group. (I) Lactate production in the control, AGEs, and AGEs+Ly294002 group. AGEs: advanced glycation end products; SASP: senescence-associated secretory phenotype; OD: optical density. *p < 0.05, **p < 0.01, and ***p < 0.001.
To further explore the role of AKT/mTOR signaling pathway activation in AGE-induced glycolysis and inflammaging, hPDLFs were pretreated with 10 mmol/L Ly294002, an inhibitor of the AKT/mTOR signaling pathway, for 2 hours, followed by stimulation with 200 µg/mL AGEs for 24 hours. The activation of the AKT/mTOR signaling pathway, glycolysis, and inflammaging-related changes were measured, and the results are presented in Figure 4. The ratios of p-AKT/AKT and p-mTOR/mTOR were significantly reduced in hPDLFs pretreated with Ly294002 and stimulated with AGEs compared to those exposed to AGEs alone, confirming that Ly294002 inhibits the activation of the AKT/mTOR signaling pathway. Moreover, the levels of HKII, PKM2, GLUT1, LDHA, as well as lactate production and glucose uptake, were significantly decreased after Ly294002 pretreatment and AGE stimulation, indicating that inhibition of the AKT/mTOR signaling pathway downregulated AGE-induced glycolysis. Additionally, Ly294002 pretreatment significantly reduced the expression levels of SASP factors, as well as p16, p21, p53, and β-galactosidase activity, suggesting that inhibiting the activation of the AKT/mTOR signaling pathway alleviated AGE-induced inflammaging.
Block of RAGE-inhibited AGE-induced activation of AKT/mTOR pathway, glycolysis, and inflammaging
hPDLFs were treated with 200 µg/mL AGEs for 24 hours, and the expression level of the RAGE was assessed via WB. The results, shown in Figure 5, indicate that exposure to 200 µg/mL AGEs resulted in a noticeable increase in RAGE expression. Moreover, when 500 nmol/L FPS-ZM1, an antagonist of RAGE, was added to block RAGE, the AGE-induced increase in RAGE expression was significantly suppressed, suggesting that AGEs induce RAGE expression in a positive feedback manner.
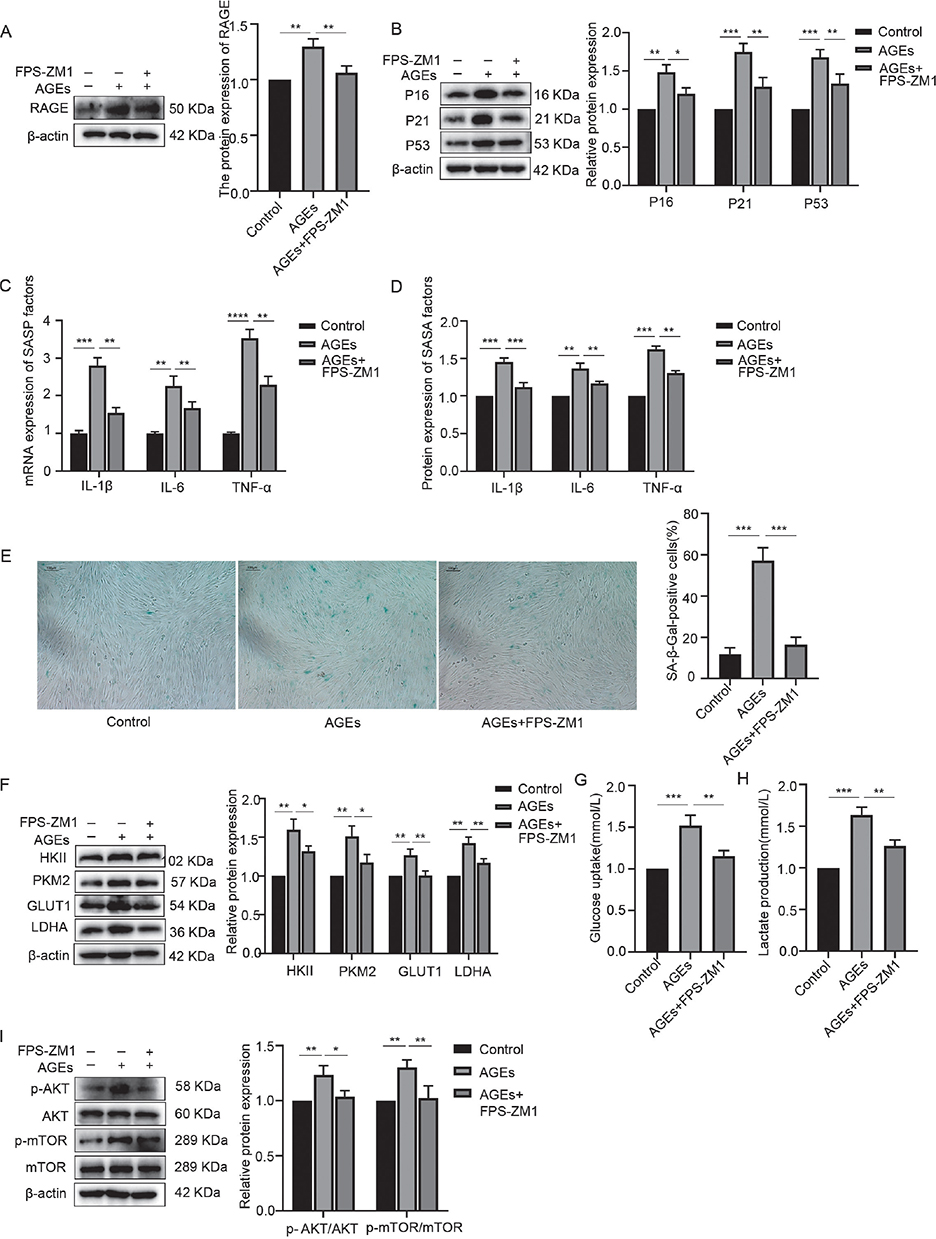
Figure 5. Block of RAGE-inhibited AGE-induced activation of AKT/mTOR pathway, glycolysis, and inflammaging. (A) RAGE expression in the control, AGEs, and AGEs+FPS-ZM1 group. (B) Western blotting of senescence characteristic protein of p16, p21, p53 in the control, AGEs, and AGEs+FPS-ZM1 group. (C) mRNA expression of SASP factors including IL-1β, IL-6, and TNF-α in the control, AGEs, and AGEs+FPS-ZM1 group. (D) Protein expression of SASP factors including IL-1β, IL-6, TNF-α in the control, AGEs, and AGEs+FPS-ZM1 group. (E) SA-β-Gal staining in the control, AGEs, and AGEs+FPS-ZM1 group. (F) Relative OD value of SA-β-Gal staining in each group. (G) Western blotting of key enzymes of glycolysis including HKII, PKM2, GLUT1, LDHA in the control, AGEs, and AGEs+FPS-ZM1 group. (H) Glucose uptake in the control, AGEs, and AGEs+FPS-ZM1 group. (I) Lactate production in the control, AGEs, and AGEs+FPS-ZM1 group. (J) Western blotting of AKT, p-AKT, mTOR, p-mTOR in the control, AGEs, and AGEs+FPS-ZM1 group. RAGE: receptor for advanced glycation end products; AGEs: advanced glycation end products; SASP: senescence-associated secretory phenotype; OD: optical density. *p < 0.05, **p < 0.01, and ***p < 0.001.
To further investigate the role of RAGE in AGE-induced changes in hPDLFs, 500 nmol/L FPS-ZM1 was used to block RAGE, followed by stimulation with 200 µg/mL AGEs for 24 hours. The results, shown in Figure 5, revealed that compared to the AGE stimulation group, the levels of inflammaging-related markers, glycolysis-related markers, and phosphorylation of AKT and mTOR were significantly reduced in the AGEs plus FPS-ZM1 group. This indicates that blocking RAGE inhibited the AGE-induced activation of the AKT/mTOR pathway, glycolysis, and inflammaging.
AGEs significantly promote lipopolysaccharide-induced secretion of inflammatory factors
hPDLFs were pretreated with 200 µg/mL AGEs for 12 hours and then stimulated with lipopolysaccharide (LPS) for 24 hours, after which the secretion of pro-inflammatory cytokines was measured. The results, shown in Figure 6, indicate that pro-inflammatory cytokine secretion was significantly higher in the group co-treated with AGEs and LPS compared to the group with LPS stimulation alone, suggesting that AGEs promote LPS-induced inflammation.
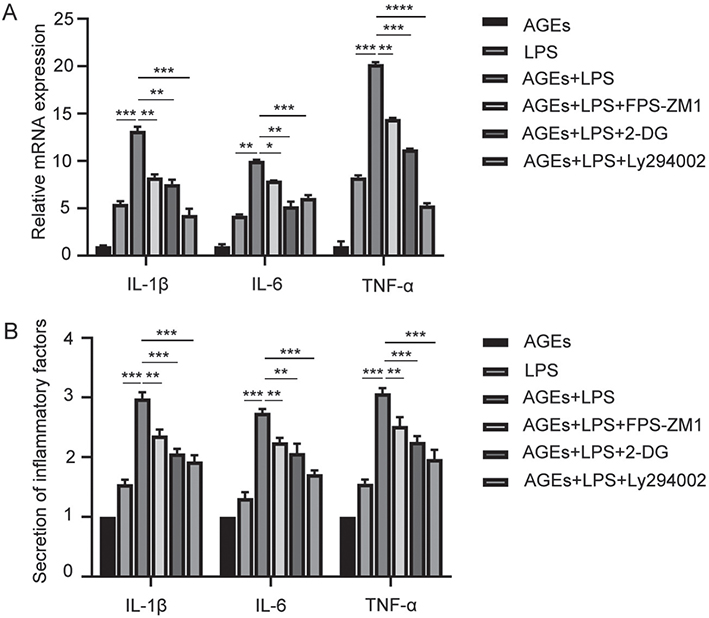
Figure 6. AGEs significantly promote LPS-induced secretion of inflammatory factors. (A) mRNA expression of inflammatory factors including IL-1β, IL-6, and TNF-α in the AGEs, LPS, AGEs+LPS, AGEs+LPS+FPS-ZM1, AGEs+LPS+2-DG, and AGEs+LPS+Ly294002 group. (B) Secretion of inflammatory factors including IL-1β, IL-6, TNF-α in the AGEs, LPS, AGEs+LPS, AGEs+LPS+FPS-ZM1, AGEs+LPS+2-DG, and AGEs+LPS+Ly294002 group. AGEs: advanced glycation end products; LPS: lipopolysaccharide; 2-DG: 2-deoxyglucose. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
To further explore the possible mechanism underlying the promotion of LPS-induced inflammation by AGEs, FPS-ZM1, LY294002, and 2-DG were co-administered with AGEs plus LPS. All three inhibitors downregulated the pro-inflammatory cytokine secretion induced by AGEs and LPS (Figure 6), indicating that RAGE, the AKT/mTOR pathway, and glycolysis are involved in the AGE-mediated promotion of LPS-induced inflammation.
Discussion
In this study, we found that AGEs could induce inflammaging, which may be linked to the increased inflammatory response in hPDLFs stimulated by LPS. We also uncovered the roles of glycolysis and the RAGE/AKT/mTOR signaling pathway in AGE-induced inflammaging. These findings highlight the involvement of inflammaging in the AGE-enhanced inflammation triggered by LPS, potentially offering new strategies for the treatment of periodontitis aggravated by AGEs.
Periodontitis is an age-related disease, and inflammaging has been shown to be closely associated with its progression [3]. Previous studies have demonstrated that inflammaging-induced epithelial barrier dysfunction plays a key role in the exacerbation of periodontitis, particularly in the context of diabetes [14]. In terms of inflammaging, the characteristic proteins of senescence, SASP factors, and positive staining for SA-β-gal are considered the best markers for identifying inflammaging in cells [23]. The periodontal ligament is a critical site for the host’s immune response to external pathogen stimulation [24], with periodontal ligament fibroblasts (PDLFs) playing an essential role in this process. Overexpression of pro-inflammatory cytokines in PDLFs can amplify local inflammatory responses and mediate periodontal tissue damage by continuously triggering immune responses [25].
In this study, hPDLFs were stimulated with AGEs, and we found that AGEs promoted the expression of characteristic senescence proteins, including p16, p21, and p53, SASP factors such as IL-1β, IL-6, and TNF-α, and increased the number of cells with positive SA-β-gal staining. Antagonizing AGEs binding to RAGE downregulated these inflammaging markers, indicating that AGEs can induce inflammaging in hPDLFs. As periodontitis is triggered by bacterial infection, with LPS being a major virulence factor in periodontitis, LPS stimulation is commonly used as an in vitro model to study periodontitis [26]. In this study, hPDLFs were pretreated with AGEs and then stimulated with LPS to simulate the role of AGEs in periodontitis. The results showed that AGEs enhanced the expression of pro-inflammatory cytokines induced by LPS. Collectively, these findings suggest that AGEs can induce inflammaging and exacerbate periodontitis, with inflammaging potentially serving as the mechanism by which AGEs aggravate the disease.
The molecular mechanisms underlying AGE-induced inflammaging in hPDLFs remain unclear. Recent studies have shown that senescence is often accompanied by alterations in energy metabolism [17, 27], with glycolysis, a key energy metabolism pathway, being closely linked to SASP secretion and cellular senescence [22, 28]. Evidence suggests that PKM2, a rate-limiting enzyme in glycolysis, plays a role in SASP and senescence [21, 29], and that inhibiting glycolysis can reduce β-galactosidase activity and suppress senescence [30]. On the other hand, in γ-ray-induced senescent cells, the glycolytic flux is significantly elevated [31]. The pro-inflammatory cytokine IL-6, a component of the SASP, directs glucose metabolism toward glycolysis [32]. Additionally, TNF-α, another important SASP component, induces metabolic reprogramming that favors glycolytic activity [33]. Collectively, these findings suggest that glycolysis promotes cellular senescence, and senescence, in turn, reinforces glycolytic metabolism. In this study, we found that AGEs increased glucose uptake and lactate production, along with the expression levels of PKM2 and other key glycolytic enzymes, including HKII, GLUT1, and LDHA. These results were consistent with previous studies [20] and suggest that AGEs shift cellular metabolism toward glycolysis. To further investigate the role of glycolysis in AGE-induced inflammaging, we co-administered 2-DG, a glycolysis inhibitor, with AGEs. The results showed that inhibiting glycolysis attenuated the expression levels of inflammaging-related factors induced by AGEs. These findings indicate that AGEs induce inflammaging by promoting glycolysis. Further studies are warranted to comprehensively evaluate the role of inflammaging in AGE-induced glycolysis promotion, and the interplay between glycolysis and inflammaging may influence the identification of potential therapeutic targets for AGE-related diseases.
However, the mechanism by which AGEs promote glycolysis remains unclear. Many studies have demonstrated that the AKT/mTOR signaling pathway plays a crucial role in regulating glycolysis [34, 35], with inhibition and over-activation of the AKT/mTOR pathway suppressing and promoting glycolysis, respectively [36, 37]. The results of this study showed that AGEs could phosphorylate AKT and mTOR to activate the AKT/mTOR signaling pathway. Inhibition of AKT/mTOR signaling by Ly294002 reduced glycolysis and inflammaging induced by AGEs, confirming that the AKT/mTOR pathway is involved in AGE-induced glycolysis and inflammaging. Finally, a RAGE antagonist was used to explore the role of RAGE in AGE-induced inflammaging, glycolysis, and activation of the AKT/mTOR signaling pathway. As is well known, AGEs primarily regulate cell metabolism by binding to RAGE [7]. The results of this study showed that the RAGE antagonist could downregulate the activation of the AKT/mTOR pathway, glycolysis, and inflammaging, suggesting that AGEs bind to RAGE to activate the AKT/mTOR pathway, increase glycolysis, and induce inflammaging.
In summary, it can be concluded that AGEs bind to RAGE to activate the AKT/mTOR signaling pathway, which shifts cell metabolism toward glycolysis and ultimately induces in-flammaging. At the same time, AGEs enhance the inflammation induced by LPS. We propose a potential link between inflammaging and the increased inflammation in periodontitis aggravated by AGEs. To date, there are few therapeutic strate-gies available for AGE-aggravated periodontitis. Therefore, the findings of this study provide an experimental basis for the prevention and treatment of AGE-aggravated periodontitis by targeting inflammaging through the RAGE/AKT/mTOR pathway and glycolysis. However, further research is needed to fully elucidate the specific mechanisms by which AGEs contribute to periodontitis.
Conclusion
Our study indicated that AGEs induced inflammaging through binding to RAGE to activate AKT/mTOR pathway and eventually enhancing glycolysis level, which may contribute to the increased inflammatory response triggered by LPS. These findings suggest that inflammaging is a critical mechanism through which AGEs exacerbate periodontitis.
Authors’ contributions
XL and SJ wrote and edited the manuscript. SJ, XL, MM, WC, LY, WH, LJ, and LW performed the experiments. SJ, LY, and SJ analyzed the data. SJ, DQ, and CH conceived and designed the study. All authors reviewed and approved the final manuscript. All authors read and approved the final manuscript. The authors declare that they have no conflicts of interest.
Ethical approval
The study was approved by the Ethics Committee of the School of Stomatology at Guizhou Medical University, Ethical Review Document No. 31 of 2021.
Informed consent
Patients enrolled in the study were aged 18–25 years and provided signed informed consent.
References
[1] Slots J. Periodontitis: facts, fallacies and the future. Periodontol 2000. 2017;75:7–23. https://doi.org/10.1111/prd.12221
[2] Kassebaum NJ, Bernabé E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res. 2014;93:1045–53. https://doi.org/10.1177/0022034514552491
[3] Genco RJ, Sanz M. Clinical and public health implications of periodontal and systemic diseases: an overview. Periodontol 2000. 2020;83:7–13. https://doi.org/10.1111/prd.12344
[4] Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, Makrilakis K, et al. Periodontitis and diabetes: a two-way relationship. Diabetologia. 2012;55:21–31. https://doi.org/10.1007/s00125-011-2342-y
[5] Marruganti C, Suvan JE, D’Aiuto F. Periodontitis and metabolic diseases (diabetes and obesity): tackling multimorbidity. Periodontol 2000. 2023. https://doi.org/10.1111/prd.12536
[6] Chopra A, Jayasinghe TN, Eberhard J. Are inflamed periodontal tissues endogenous source of advanced glycation end-products (AGEs) in individuals with and without diabetes mellitus? A systematic review. Biomolecules. 2022;12:642. https://doi.org/10.3390/biom12050642
[7] Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced glycation end-products (AGEs): formation, chemistry, classification, receptors, and diseases related to AGEs. Cells. 2022;11:1312. https://doi.org/10.3390/cells11081312
[8] Sonnenschein SK, Meyle J. Local inflammatory reactions in patients with diabetes and periodontitis. Periodontol 2000. 2015;69:221–54. https://doi.org/10.1111/prd.12089
[9] Baima G, Romandini M, Citterio F, Romano F, Aimetti M. Periodontitis and accelerated biological aging: a geroscience approach. J Dent Res. 2022;101:125–32. https://doi.org/10.1177/00220345211037977
[10] Clark D, Radaic A, Kapila Y. Cellular mechanisms of inflammaging and periodontal disease. Front Dent Med. 2022;3:844865. https://doi.org/10.3389/fdmed.2022.844865
[11] Prattichizzo F, De Nigris V, La Sala L, Procopio AD, Olivieri F, Ceriello A. ‘Inflammaging’ as a druggable target: a senescence-associated secretory phenotype-centered view of type 2 diabetes. Oxid Med Cell Longev. 2016;2016:1810327. https://doi.org/10.1155/2016/1810327
[12] O’Sullivan O, Ladlow P, Steiner K, Hillman C, Stocks J, Bennett AN, et al. Current status of catabolic, anabolic and inflammatory biomarkers associated with structural and symptomatic changes in the chronic phase of post-traumatic knee osteoarthritis – a systematic review. Osteoarthr Cartil Open. 2023;5:100412. https://doi.org/10.1016/j.ocarto.2023.100412
[13] Nandakumar KS, Fang Q, Wingbro Ågren I, Bejmo ZF. Aberrant activation of immune and non-immune cells contributes to joint inflammation and bone degradation in rheumatoid arthritis. Int J Mol Sci. 2023;24:15883. https://doi.org/10.3390/ijms242115883
[14] Zhang P, Lu B, Zhu R, Yang D, Liu W, Wang Q, et al. Hyperglycemia accelerates inflammaging in the gingival epithelium through inflammasomes activation. J Periodontal Res. 2021;56:667–78. https://doi.org/10.1111/jre.12863
[15] Albuquerque-Souza E, Crump KE, Rattanaprukskul K, Li Y, Shelling B, Xia-Juan X, et al. TLR9 mediates periodontal aging by fostering senescence and inflammaging. J Dent Res. 2022;101:1628–36. https://doi.org/10.1177/00220345221110108
[16] Shen CY, Li KJ, Wu CH, Lu CH, Kuo YM, Hsieh SC, et al. Unveiling the molecular basis of inflamm-aging induced by advanced glycation end products (AGEs)-modified human serum albumin (AGE-HSA) in patients with different immune-mediated diseases. Clin Immunol. 2023;252:109655. https://doi.org/10.1016/j.clim.2023.109655
[17] Giuliani A, Giudetti AM, Vergara D, Del Coco L, Ramini D, Caccese S, et al. Senescent endothelial cells sustain their senescence-associated secretory phenotype (SASP) through enhanced fatty acid oxidation. Antioxidants (Basel). 2023;12:1956. https://doi.org/10.3390/antiox12111956
[18] Song MJ, Park CH, Kim H, Han S, Lee SH, Lee DH, et al. Carnitine acetyltransferase deficiency mediates mitochondrial dysfunction- induced cellular senescence in dermal fibroblasts. Aging Cell. 2023;22:e14000. https://doi.org/10.1111/acel.14000
[19] Tewari D, Patni P, Bishayee A, Sah AN, Bishayee A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: a novel therapeutic strategy. Semin Cancer Biol. 2022;80:1–17. https://doi.org/10.1016/j.semcancer.2019.12.008
[20] Atawia RT, Batori RK, Jordan CR, Kennard S, Antonova G, Bruder-Nascimento T, et al. Type 1 diabetes impairs endothelium-dependent relaxation via increasing endothelial cell glycolysis through advanced glycation end products, PFKFB3, and Nox1-mediated mechanisms. Hypertension. 2023;80:2059–71. https://doi.org/10.1161/hypertensionaha.123.21341
[21] Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–54. https://doi.org/10.1084/jem.20150900
[22] Liao EC, Hsu YT, Chuah QY, Lee YJ, Hu JY, Huang TC, et al. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis. 2014;5:e1255. https://doi.org/10.1038/cddis.2014.220
[23] Kruglov V, Jang IH, Camell CD. Inflammaging and fatty acid oxidation in monocytes and macrophages. Immunometabolism (Cobham). 2024;6:e00038. https://doi.org/10.1097/in9.0000000000000038
[24] Ikegami K, Yamashita M, Suzuki M, Nakamura T, Hashimoto K, Kitagaki J, et al. Cellular senescence with SASP in periodontal ligament cells triggers inflammation in aging periodontal tissue. Aging (Albany NY). 2023;15:1279–305. https://doi.org/10.18632/aging.204569
[25] Naruishi K. Biological roles of fibroblasts in periodontal diseases. Cells. 2022;11:3345. https://doi.org/10.3390/cells11213345
[26] Suh HN, Ji JY, Heo JS. Translating proteome and transcriptome dynamics of periodontal ligament stem cell-derived secretome/conditioned medium in an in vitro model of periodontitis. BMC Oral Health. 2024;24:390. https://doi.org/10.1186/s12903-024-04167-z
[27] Kobayashi H, Yoshimoto C, Matsubara S, Shigetomi H, Imanaka S. Altered energy metabolism, mitochondrial dysfunction, and redox imbalance influencing reproductive performance in granulosa cells and oocyte during aging. Reprod Sci. 2024;31:906–16. https://doi.org/10.1007/s43032-023-01394-7
[28] Marrocco A, Ortiz LA. Role of metabolic reprogramming in pro- inflammatory cytokine secretion from LPS or silica-activated macrophages. Front Immunol. 2022;13:936167. https://doi.org/10.3389/fimmu.2022.936167
[29] Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129–34. https://doi.org/10.1073/pnas.1014769108
[30] Murao N, Yokoi N, Takahashi H, Hayami T, Minami Y, Seino S. Increased glycolysis affects β-cell function and identity in aging and diabetes. Mol Metab. 2022;55:101414. https://doi.org/10.1016/j.molmet.2021.101414
[31] James EL, Michalek RD, Pitiyage GN, de Castro AM, Vignola KS, Jones J, et al. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J Proteome Res. 2015;14:1854–71. https://doi.org/10.1021/pr501221g
[32] Xu S, Deng KQ, Lu C, Fu X, Zhu Q, Wan S, et al. Interleukin-6 classic and trans-signaling utilize glucose metabolism reprogramming to achieve anti- or pro-inflammatory effects. Metabolism. 2024;155:155832. https://doi.org/10.1016/j.metabol.2024.155832
[33] Manosalva C, Alarcon P, Quiroga J, Teuber S, Carretta MD, Bustamante H, et al. Bovine tumor necrosis factor-alpha increases IL-6, IL-8, and PGE2 in bovine fibroblast-like synoviocytes by metabolic reprogramming. Sci Rep. 2023;13:3257. https://doi.org/10.1038/s41598-023-29851-y
[34] Chen S, Tao Y, Wang Q, Ren J, Jing Y, Huang J, et al. Glucose induced-AKT/mTOR activation accelerates glycolysis and promotes cell survival in acute myeloid leukemia. Leuk Res. 2023;128:107059. https://doi.org/10.1016/j.leukres.2023.107059
[35] Chen L, Li X, Deng Y, Chen J, Huang M, Zhu F, et al. The PI3K-Akt-mTOR pathway mediates renal pericyte-myofibroblast transition by enhancing glycolysis through HKII. J Transl Med. 2023;21:323. https://doi.org/10.1186/s12967-023-04167-7
[36] Wang K, Li J, Zhou B. KIAA0101 knockdown inhibits glioma progression and glycolysis by inactivating the PI3K/AKT/mTOR pathway. Metab Brain Dis. 2022;37:489–99. https://doi.org/10.1007/s11011-021-00863-9
[37] Duggan MR, Weaver M, Khalili K. PAM (PIK3/AKT/mTOR) signaling in glia: potential contributions to brain tumors in aging. Aging (Albany NY). 2021;13:1510–27. https://doi.org/10.18632/aging.202459