ORIGINAL ARTICLE
Germline variants in patients diagnosed with pediatric soft tissue sarcoma
Synnøve Yndestada,b , Hans Kristian Hauglandc, Dorota Goplenb, Dorota Wojcikd, Stian Knappskoga,b and Per Eystein Lønninga,b
, Hans Kristian Hauglandc, Dorota Goplenb, Dorota Wojcikd, Stian Knappskoga,b and Per Eystein Lønninga,b
aK.G. Jebsen Center for Genome-Directed Cancer Therapy, Department of Clinical Science, University of Bergen, Bergen, Norway; bDepartment of Oncology, Haukeland University Hospital, Bergen, Norway; cDepartment of Pathology, Haukeland University Hospital, Bergen, Norway; dDepartment of Pediatrics, Haukeland University Hospital, Bergen, Norway
ABSTRACT
Background: While soft tissue sarcomas affect younger patients, few studies have assessed the distribution of underlying pathogenic germline variants.
Patients and methods: We retrospectively identified all pediatric and young adult patients (0–22 years) at Haukeland University Hospital, Norway (1981–2019), through clinical and pathological records. We identified n = 46 eligible patients. From these 46 patients, adequate material representing normal tissue was available for n = 41 cases (n = 24 diagnosed with rhabdomyosarcoma, 9 with synovial sarcomas, 2 with Ewing sarcomas, and 6 without further classification), with matching tumor tissue for n = 40. Normal tissue samples were analyzed for germline pathogenic variants (PVs) by targeted sequencing of 360 cancer genes.
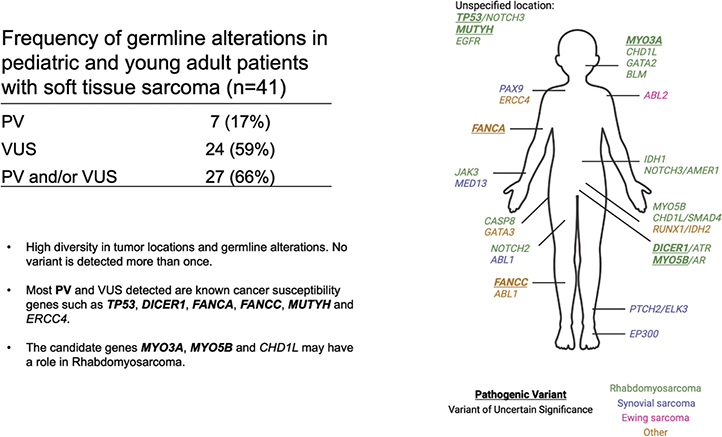
Results: Out of the 41 analyzed cases, we found PVs or likely PVs in 7 (17%). These variants were found in TP53, MUTYH, FANCC, DICER1, FANCA, MYO3A, and MYO5B. Supporting the causality of these PVs, four cases revealed loss of heterozygosity (LOH) of the wild-type allele in the tumor tissue, one patient with a PV in DICER1 had a second somatic variant in DICER1, and a patient with a PV in TP53 had the altered allele amplified in the tumor. For three out of five with available family history, a history of other cancers in relatives was recorded. Among genes with variants of uncertain significance, CHD1L was of particular interest, revealing a stop-gain and a missense variant.
Interpretation: A high fraction of young patients with soft tissue sarcoma harbor PVs. Among the genes affected, we substantiate a potential role of MYO5B and propose a potential role for MYO3A.
KEYWORDS: Soft tissue sarcoma; pathogenic germline variants; hereditary; MYO3A; MYO5B; CHD1L
Citation: ACTA ONCOLOGICA 2024, VOL. 63, 586–591. https://doi.org/10.2340/1651-226X.2024.40730.
Copyright: © 2024 The Author(s). Published by MJS Publishing on behalf of Acta Oncologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), allowing third parties to copy and redistribute the material in any medium or format and to remix, transform, and build upon the material, with the condition of proper attribution to the original work.
Received: 6 May 2024; Accepted: 28 June 2024; Published: 22 July 2024
CONTACT Stian Knappskog stian.knappskog@uib.no K.G. Jebsen Center for Genome-Directed Cancer Therapy, Department of Clinical Science, University of Bergen, Bergen, Norway; Department of Oncology, Haukeland University Hospital, Bergen, Norway
Supplemental data for this article can be accessed online at https://doi.org/10.2340/1651-226X.2024.40730
Competing interests and funding: The authors report there are no competing interests to declare.
Introduction
Sarcoma is a highly diverse group of cancers originating from mesenchymal cells in the bones or soft tissues. While sarcomas make up less than 1% of all adult solid malignant cancers, they account for more than 20% of solid malignant cancers in the pediatric population [1]. Some pediatric solid tumors, like retinoblastomas and Wilms tumors, are strongly linked to defined monogenetic germline variants but for the majority of pediatric cancers, including sarcomas, the contribution of germline alterations is less well defined [2].
Rhabdomyosarcoma (RMS) is the soft tissue sarcoma most frequently diagnosed in childhood [3]. There are two major subtypes. Embryonal RMSs accounting for about 70% of cases has a peak incidence around 2 to 4 years of age. The alveolar subtype, characterized by frequent translocations between PAX3 or PAX7 and FOXO1 genes, is diagnosed in children and young adults up to 20 years of age with no distinct age peak [4, 5]. While RMS has been associated with rare cancer-disposition syndromes such as the Li–Fraumeni syndrome and neurofibromatosis type-1, these syndromes account for a limited number of cases [6]. Recent studies have estimated the fraction of RMSs associated with pathogenic germline variants to be less than 10% [7, 8].
Searching for potential cancer-predisposing germline variants in childhood cancers provides several challenges. These tumors are extremely rare; thus, even assuming a relative odds ratio of > 50, similar to BRCA1 and breast cancer risk [9], the likelihood of detecting several cases of pediatric/adolescent malignancies in a family is small. Then, before the time of modern treatment, most children diagnosed with a pediatric solid tumor died before reaching age of reproduction, limiting the possibility of identifying germline aberrations in familial linkage studies. A remaining feasible strategy is to assess germline status in individual patients and, after detection of potentially pathogenic variants, identify additional indications of causality, for example, LOH in tumors.
Conflicting evidence has linked congenital malformations to risk of pediatric cancers [10–14]. During screening of potential germline variants in a 20-year-old man with RMS located in the prostate, we identified a germline variant in the CHD1L gene (amplified in liver cancer 1; ALC1), encoding a chromodomain helicase in which mutations have been associated with urinary tract anomalies [15, 16]. Triggered by this finding, we initiated the present study to screen for germline variants across archive samples from young patients diagnosed with soft tissue sarcomas to potentially detect novel risk variants for these malignancies.
Patients and methods
Patients
Pediatric and young adult (0–22 years) patients, treated for soft tissue sarcomas, in our institution between year 1981–2019 was retrospectively identified through hospital (clinical and pathological) records. We identified n = 46 eligible patients with pathological confirmed diagnosis of soft tissue sarcoma and availability biomaterial in our diagnostic biobank. From the 46 patients, adequate material representing normal tissue was available for n = 41 cases, with matching tumor tissue for n = 40.
Ethics
The protocol was approved by the Regional Ethical Committee of Western Norway (REK vest 50564.2019).
DNA extraction, library prep, and sequencing
Normal and tumor tissue samples were collected from FFPE blocks by extracting 0.8–1 mM core (5 mg tissue). DNA was isolated by adaptive focused acoustics (AFA)-based extraction using the Covaris truXTRAC FFPE DNA kit (Woburn, MA, USA) as previously described (Supplementary Information and [17]).
DNA (normal and tumor samples) was analyzed by targeted massive parallel sequencing of a 360 cancer gene panel, as previously described [18].
Classification of germline variants
After sequencing and initial processing using the local run manager software (Illumina MiSeq instrument), variants in known cancer predisposition genes were classified according to the ACMG criteria [19] using the ‘Cancer Predisposition Sequencing Reporter’ (CPSR) module within the python package ‘The Personal Cancer Genome Reporter’ (PCGR) v 1.0.3 [20] or by hard filtering and manual curation for genes not covered by PCGR (see Supplementary Information).
Somatic variant calling
Alignment was performed using MiSeq reporter against UCSC hg19, and functional annotation was performed by Annovar [21]. For the matched tumor-normal pairs, mutations and small indels were called by CaVEman [22] and Pindel [23], respectively. Copy number analysis on matched tumor and normal tissue was performed using FACET (https://github.com/mskcc/facets) [24].
Results
For the period between 1981 and 2019, 46 pediatric, teenage, and young adult patients treated for soft tissue sarcoma at Haukeland University Hospital were identified. The age range was 0–22 years; 22 cases were female while 24 were male. Adequate normal tissue was available for 41 cases, and matched tumor-normal pairs was available for 40 cases (Table 1, Supplementary Data 2).
As for histopathology, 24 tumors (59%) were classified as RMSs, 9 as synovial sarcoma, two Ewing sarcomas affecting soft tissue, while the remaining 6 were sarcomas without further classification (Table 1). Genetic alterations in the tumors are summarized in Supplementary Figure 2. The most frequently somatically mutated genes were NRAS, FGFR4, and CTNNB1, which are all commonly mutated in RMSs [25].
Germline pathogenic variants
Out of the 41 analyzed cases, we found germline pathogenic or likely pathogenic germline variants (PV) in 7 cases (17%; Figure 1; Table 2). Among the detected variants, 5 were in known cancer predisposition genes (TP53, MUTYH, FANCC, DICER1, and FANCA), while one patient harbored a variant in MYO3A, and another one in MYO5B. Both the latter genes belong to the myosin family, and functional loss of MYO3A and MYO5B disrupts cell polarity and causes malformation of cochlea hairs and microvillus leading to deafness and microvillus inclusion disease, respectively [26, 27].
| Sample ID | Gene | Chr. | Position | Ref. | Alt. | Variant Class | AA Change | Variant Call | VAF blood | VAF tumor | Variant CNA1 | Cancer type | Subtype | Fusion status | Primary site | Age | Sex | Familial cancer2 |
| 017n | TP53 | chr17 | 7579418 | G | GA | Frame shift | p.S90fs | Pathogenic | 0.4000 | 0.5965 | Amp | Rhab. | Embryonal | NA | NA | 7 | Male | Y |
| 041n | DICER1 | chr14 | 95578563 | G | A | Nonsense | p.R688X | Pathogenic | 0.4850 | 0.4897 | NA3 | Rhab. | Embryonal | NA | Gynecologic | 15 | Female | Y |
| 022n | MUTYH | chr1 | 45799108 | G | A | Missense | p.R106W | Likely pathogenic | 0.4302 | 0.9256 | LOH | Rhab. | Alveolar | NA | NA | 7 | Male | N |
| 033n | MYO3A | chr10 | 26463065 | A | - | Frame shift | p.Q1291fs | Likely pathogenic | 0.5196 | 0.7045 | LOH | Rhab. | Embryonal | NA | Head and neck | 13 | Female | Unknown |
| 040n | FANCC | chr9 | 97864024 | G | A | Nonsense | p.R548X | Likely pathogenic | 0.2941 | 0.5096 | NA | uncl. | Unknown | NA | Extremities | 0 | Male | Unknown |
| 045n | MYO5B | chr18 | 47500736 | C | A | Missense | p.V436F | Likely pathogenic | 0.5043 | 0.5750 | LOH | Rhab. | Embryonal | NA | Pelvic | 1 | Female | N |
| 047n | FANCA | chr16 | 89813256 | T | C | Missense | p.T1131A | Likely pathogenic | 0.4706 | 0.5455 | LOH | uncl. | Unknown | NA | Extremities | 19 | Male | Y |
| 001n | NOTCH2 | chr1 | 120497768 | C | T | Missense | p.R705H | VUS | 0.3947 | 0.4310 | No | Rhab. | Alveolar | positive | Extremities | 7 | Male | NA |
| 002n | PTCH2 | chr1 | 45297405 | T | C | Missense | p.K197R | VUS | 0.4865 | 0.4360 | No | Sync. | Unknown | NA | Extremities | 6 | Female | NA |
| 002n | ELK3 | chr12 | 96653576 | G | A | Missense | p.S357N | VUS | 0.5000 | 0.5031 | No | Sync. | Unknown | NA | Extremities | 6 | Female | NA |
| 005n | CASP8 | chr2 | 202149811 | G | C | Missense | p.A418P | VUS | 0.4769 | 0.3827 | No | Rhab. | Alveolar | NA | Trunk | 9 | Female | NA |
| 006n | ABL2 | chr1 | 179090902 | G | A | Missense | p.A263V | VUS | 0.5102 | 0.5294 | No | Ew. | Unknown | NA | Trunk | 17 | Male | NA |
| 007n | NOTCH3 | chr19 | 15276251 | G | A | Missense | p.R1915C | VUS | 0.5085 | 0.5366 | No | Rhab. | Embryonal | NA | Abdominal | 2 | Female | NA |
| 007n | AMER1 | chrX | 63412159 | G | C | Missense | p.D336E | VUS | 0.4125 | 0.4974 | NA | Rhab. | Embryonal | NA | Abdominal | 2 | Female | NA |
| 009n | CHD1L | chr1 | 146743877 | G | A | Missense | p.R402K | VUS | 0.3784 | 0.1036 | No | Rhab. | Alveolar | PAX3 | Head and neck | 12 | Male | NA |
| 010n | IDH1 | chr2 | 209108301 | T | C | Missense | p.Y183C | VUS | 0.4519 | 0.3281 | No | Rhab. | Embryonal | PAX3 | Abdominal | 6 | Female | NA |
| 013n | SMAD4 | chr18 | 48575164 | G | T | Missense | p.D120Y | VUS | 0.4667 | 0.4403 | NA | Rhab. | Unknown | inconclusive | Genitourinary | 20 | Male | Y |
| 013n | CHD1L | chr1 | 146766153 | C | T | Nonsense | p.R857X | VUS | 0.4652 | 0.4138 | No | Rhab. | Unknown | inconclusive | Genitourinary | 20 | Male | Y |
| 014n | MYO5B | chr18 | 47431169 | G | A | Missense | p.A815V | VUS | 0.5083 | 0.5446 | NA | Rhab. | Alveolar | PAX7 | Genitourinary | 3 | Female | NA |
| 016n | EGFR | chr7 | 55259485 | C | T | Missense | p.P848L | VUS | 0.5507 | 0.4032 | No | Rhab. | Pleomorph | NA | NA | 12 | Male | N |
| 017n | NOTCH3 | chr19 | 15302615 | C | G | Missense | p.G248A | VUS | 0.4837 | 0.5217 | No | Rhab. | Embryonal | NA | NA | 7 | Male | Y |
| 018n | GATA3 | chr10 | 8097700 | C | A | Missense | p.H28N | VUS | 0.4854 | 0.9239 | LOH | uncl. | Undifferentiated | NA | Trunk | 11 | Female | NA |
| 020n | BLM | chr15 | 91312417 | C | A | Missense | p.L788I | VUS | 0.4300 | 0.4387 | NA | Rhab. | Embryonal | NA | Head and neck | 4 | Male | NA |
| 023n | GATA2 | chr3 | 128205754 | G | C | Missense | p.P41A | VUS | 0.4367 | 0.4789 | NA | Rhab. | Unknown | NA | Head and neck | 14 | Male | NA |
| 030n | EP300 | chr22 | 41574340 | AACC AGTT CCAGC |
A | In frame del | p.N2209_ Q2213delinsK |
VUS | 0.2961 | 0.2110 | NA | Sync. | Unknown | NA | Extremities | 20 | Female | NA |
| 032n | PAX9 | chr14 | 37132699 | T | A | Missense | p.I201N | VUS | 0.5526 | 0.4936 | NA | Sync. | Unknown | NA | Trunk | 19 | Male | NA |
| 035n | ABL1 | chr9 | 133730199 | C | T | Missense | p.R89W | VUS | 0.3929 | 0.4177 | No | Sync. | Unknown | NA | Extremities | 20 | Female | NA |
| 036n | MED13 | chr17 | 60023885 | G | A | Missense | p.P2157S | VUS | 0.4985 | 0.5111 | No | Sync. | Unknown | NA | Extremities | 5 | Female | NA |
| 037n | ERCC4 | chr16 | 14041606 | T | C | Missense | p.L718P | VUS | 0.4375 | 0.5128 | NA | uncl. | Unknown | NA | Trunk | 2 | Female | NA |
| 038n | RUNX1 | chr21 | 36259336 | A | T | Missense | p.M52K | VUS | 0.4804 | 0.4935 | LOH | uncl. | Unknown | NA | Genitourinary | 2 | Male | NA |
| 038n | IDH2 | chr15 | 90630756 | A | G | Missense | p.W244R | VUS | 0.4561 | 0.4600 | No | uncl. | Unknown | NA | Genitourinary | 2 | Male | NA |
| 040n | ABL1 | chr9 | 133738171 | C | T | Missense | p.R191C | VUS | 0.4211 | 0.4783 | NA | uncl. | Unknown | NA | Extremities | 0 | Male | Unknown |
| 041n | ATR | chr3 | 142231224 | C | A | Missense | p.S1577I | VUS | 0.4750 | 0.4605 | No | Rhab. | Embryonal | NA | Gynecologic | 15 | Female | Y |
| 045n | AR | chrX | 66766412 | C | T | Missense | p.A475V | VUS | 0.6418 | 0.4286 | No | Rhab. | Embryonal | NA | Pelvic | 1 | Female | N |
| 046n | JAK3 | chr19 | 17940964 | G | T | Missense | p.L1054M | VUS | 0.5789 | 0.7353 | LOH | Rhab. | Alveolar | positive | Extremities | 2 | Male | Unknown |
| VUS: variants of uncertain significance, VAF: variant allele frequency, CNA: copy number alteration | ||||||||||||||||||
| 1NA = Not assessed, too few SNV in area to assess accurate CNA with FACET. | ||||||||||||||||||
| 2NA = Not available. Unknown = Patient journal examined, but no relevant information found. | ||||||||||||||||||
| 3Patient 41 also harbored a somatic mutation in DICER1. | ||||||||||||||||||
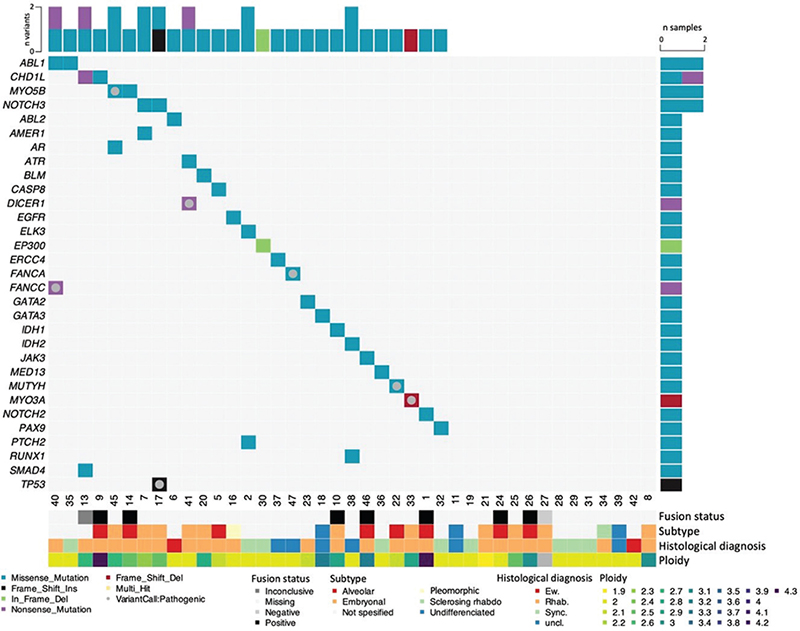
Figure 1. Germline variants in young patients with soft tissue sarcoma. Oncoplot presenting germline pathogenic variants and variants of uncertain significance (VUS) detected in selected genes (rows) in soft tissue sarcoma patients (columns). Pathogenic and likely pathogenic variants are indicated by a grey circle. The remaining variants are classified as VUS.
Regarding sarcoma subtypes, 6 of the 7 PVs were found in patients with RMS, while the last was found in patients with unclassified sarcoma (Supplementary Table 1). No pathogenic variants were discovered among individuals diagnosed with synovial sarcomas (n = 9) or Ewing sarcomas (n = 2). The presence of PVs was not biased with respect to gender, with 4 out of 7 found in males and 3 found in females. The median age of onset was 7 years among the patients with a PV, and 11 years for those with no PV detected, but this difference did not reach statistical significance (p > 0.4). The location of primary tumors in patients with a PV was diverse and did not differ from the primary sites of the tumors in patients with no PV (Table 2).
Out of the alterations classified here as PVs, the MUTYH p.R106W (dbSNP: rs765123255), FANCC p.R548X (dbSNP: rs104886457), DICER1 p.R688X (dbSNP: rs886037684), and FANCA p.T1131A (dbSNP: rs574034197) have previously been reported as pathogenic in ClinVar. Although other nucleotide changes causing TP53 S90fs have been reported the presently detected variant causing TP53 S90fs as well as the MYO3A Q1291fs and MYO5B V436F variants have not been reported in either the 1,000 genomes (1000g2014oct_all) or the gnomAD (v2.1.1) database.
Supporting the causality of the PVs with respect to tumorigenesis, 4 tumors revealed LOH with loss of the wild-type allele in the tumor tissue (Table 2; Supplementary Figure 3). For the patient with a PV in DICER1, LOH status was not assessable due to too few SNPs in the region. A second somatic variant in DICER1 was, however, found in the tumor (Figure 1, Supplementary Figures 2 and 3). Further, the patient with a PV in TP53 had the altered allele amplified in the tumor, while CNA status was unavailable for the patient with a FANCC variant.
Out of the 7 genes in which we detected PVs, 4 (TP53, DICER1, FANCC, and FANCA), have previously been reported associated with elevated risk of RMSs. The primary tumor type for those affected by PVs in these genes, in the present study, were embryonal RMS (TP53, DICER1), rhabdomyofibrosarcoma (FANCC), and osteosarcoma (FANCA). Interestingly, the tumors in patients with the novel variants in MYO3A and MYO3B, as well as MUTYH, were all RMSs. These genes have not previously been shown to be associated with elevated risk of either RMSs, Ewing sarcoma or soft tissue sarcomas in general. It should be noted that two patients in our cohort had somatic mutations in MYO3A and MYO3B (patients 17 and 33, respectively), further indicating a role for these genes in sarcoma (Supplementary Figure 2 and Supplementary Data 3).
Of the 7 patients with a PV, information about family history of cancer was available for 5. Out of these, 3 had a family history of cancer: the patients carrying the TP53, FANCA and DICER variants (Table 2; further detail in Supplementary Information).
Variants of uncertain significance
In addition to the pathogenic and likely pathogenic variants, we performed an in-depth assessment of variants of uncertain significance (VUS), in order to potentially reveal previously unknown mechanisms of early onset soft tissue sarcoma. We identified a total of 28 VUS (Figure 1; Table 2). Overall, the 41 patients harbored on average 0.7 VUS with few recurrent genes (Table 2). Among the patients harboring either a pathogenic variant or a VUS, the patients were diagnosed at a younger age (median 7 vs. 13 years) although this difference was not statistically significant.
Discussion
In the present study, we found 7 out of 41 young patients diagnosed with soft tissue sarcomas to harbor pathogenic or likely pathogenic germline variants.
In 5 out of the 7 cases with PVs, the variants were found in well-established cancer risk genes (TP53, MUTYH, FANCC, DICER1, and FANCA). Although the number of observations is small, it is interesting to note that four of these genes have critical roles in DNA repair, similar to what is found in other reports on early onset sarcoma [28, 29].
In addition to PVs in these five genes, we detected germline PVs in MYO3A and MYO5B, both involved in elongation of actin in stereocilia tips and epithelial polarization [26, 30]. Some potentially damaging germline variants in these two genes have been reported previously in different other cancer types including CNS tumors, neuroblastoma (MYO3A), and single cases of osteosarcomas and RMSs (MYO5B) [31]. A potential role of these genes to RMS evolution is further supported by our finding of somatic variants in both MYO3A and MYO5B in two different RMSs. In a study reporting germline PVs, among 1120 pediatric cancer patients [32] neither MYO3A, MYO3B, or CHD1L were covered. Thus, our present findings represent novel information about the potential role of these genes in sarcoma. Pending further validation, this may have clinical implications with respect to how variants in these genes are considered in the setting of genetic counselling.
We found VUS in several genes, where CHD1L is of particular interest. In addition to the stop-gain mutation in patient no. 013, which initiated the present study, a germline missense VUS, was detected in patient 009. Further, one patient (no.025) revealed a somatic CHD1L mutation in the tumor). Interestingly, a previous study [31] found CHD1L germline alterations among three pediatric patients diagnosed with neuroblastoma, retinoblast-oma, and a Wilms tumor, respectively.
While the pathogenic or likely pathogenic variants affected canonical cancer predisposition genes, the VUSes affected a broader repertoire of genes. To seek insight into the biology of variants observed in early onset sarcoma, we assessed the genes affected by VUSes for their association with syndromes other than cancer. Several of the genes were associated with developmental/birth defects and/or with defects in the same organ system in which the sarcomas of our cases originated. PVs in CHD1L have also been linked to congenital anomaly of kidneys and the urinary tract [15]. Another important example is MYO3A for which germline mutations are associated with deafness. Patient no 33, harbouring a pathogenic variant in MYO3A, was diagnosed with a RMS located in the soft palate. Although these observations should be seen as anecdotal observations, we believe they warrant further investigations into the potential link between genetic alterations in developmental defect syndromes and sarcomas.
In conclusion, we find a relatively high fraction of young patients with soft tissue sarcoma to harbor pathogenic or likely pathogenic germline variants. Among the genes affected by PVs, we substantiate the potential role of MYO5B and propose a potential role for MYO3A. In addition, our data suggest that CDH1L may be a candidate for further investigation with respect to risk of soft tissue sarcoma.
Author contributions
SY: Performed data analysis and interpreted results wrote the manuscript.
DG: Reviewed and identified cases provided clinical information.
DW: Reviewed and identified cases provided clinical information.
HKH: Reviewed and identified cases performed pathological review and provided clinical information.
SK: Interpreted results, provided funding, wrote the manuscript.
PEL: Conceived the study, interpreted results, provided funding, wrote the manuscript.
All authors: Approved the final version of the manuscript.
Acknowledgements
Most of the present work was performed in the Mohn Cancer Research Laboratory. The authors thank Beryl Leirvaag, Nhat Kim Duong, and Silje Bjørneklett for technical assistance. The work was funded by grants from the K.G. Jebsen foundation, The Norwegian Cancer Society and the Norwegian Health Region West.
Data availability statement
Filtered data are available in Supplementary Data 1–4. Raw data are collected, stored, and disseminated according to institutional guidelines. After publication and on formal request, raw data, including deidentified individual participant data, may be shared according to institutional procedures. Requests are via a standard pro forma describing the nature of the proposed research and extent of data requirements. Data recipients are required to enter a formal data sharing agreement, which describes the conditions for release and requirements for data transfer, storage, archiving, publication, and intellectual property. Requests are reviewed by the study team (authors of the present paper) in terms of scientific merit and ethical considerations.
References
[1] Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14. https://doi.org/10.1186/2045-3329-2-14
[2] Lu Y, Ek WE, Whiteman D, Vaughan TL, Spurdle AB, Easton DF, et al. Most common ‘sporadic’ cancers have a significant germline genetic component. Hum Mol Genet. 2014;23(22):6112–8. https://doi.org/10.1093/hmg/ddu312
[3] Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019;5:1. https://doi.org/10.1038/s41572-018-0051-2
[4] Martin-Giacalone BA, Weinstein PA, Plon SE, Lupo PJ. Pediatric rhabdomyosarcoma: epidemiology and genetic susceptibility. J Clin Med. 2021;10(9):2028. https://doi.org/10.3390/jcm10092028
[5] Arora RS, Alston RD, Eden TO, Geraci M, Birch JM. The contrasting age-incidence patterns of bone tumours in teenagers and young adults: implications for aetiology. Int J Cancer. 2012;131(7):1678–85. https://doi.org/10.1002/ijc.27402
[6] Schneider KW, Cost NG, Schultz KAP, Svihovec S, Suttman A. Germline predisposition to genitourinary rhabdomyosarcoma. Transl Androl Urol. 2020;9(5):2430–40. https://doi.org/10.21037/tau-20-76
[7] Kim J, Light N, Subasri V, Young EL, Wegman-Ostrosky T, Barkauskas DA, et al. Pathogenic germline variants in cancer susceptibility genes in children and young adults with rhabdomyosarcoma. JCO Precis Oncol. 2021;5:75–87. https://doi.org/10.1200/PO.20.00218
[8] Li H, Sisoudiya SD, Martin-Giacalone BA, Khayat MM, Dugan-Perez S, Marquez-Do DA, et al. Germline cancer predisposition variants in pediatric rhabdomyosarcoma: a report from the Children’s Oncology Group. J Natl Cancer Inst. 2021;113(7):875–83. https://doi.org/10.1093/jnci/djaa204
[9] Walsh T, Gulsuner S, Lee MK, Troester MA, Olshan AF, Earp HS, et al. Inherited predisposition to breast cancer in the Carolina Breast Cancer Study. Npj Breast Cancer. 2021;7(1):6. https://doi.org/10.1038/s41523-020-00214-4
[10] Narod SA, Hawkins MM, Robertson CM, Stiller CA. Congenital anomalies and childhood cancer in Great Britain. Am J Hum Genet. 1997;60(3):474–85.
[11] Merks JHM, Caron HN, Hennekam RCM. High incidence of malformation syndromes in a series of 1,073 children with cancer. Am J Med Genet Part A. 2005;134A(2):132–43. https://doi.org/10.1002/ajmg.a.30603
[12] Jongmans MCJ, Loeffen J, Waanders E, Hoogerbrugge PM, Ligtenberg MJL, Kuiper RP, et al. Recognition of genetic predisposition in pediatric cancer patients: an easy-to-use selection tool. Eur J Med Genet. 2016;59(3):116–25. https://doi.org/10.1016/j.ejmg.2016.01.008
[13] Yang P, Grufferman S, Khoury MJ, Schwartz AG, Kowalski J, Ruymann FB, et al. Association of childhood rhabdomyosarcoma with neurofibromatosis type I and birth defects. Genet Epidemiol. 1995;12(5):467–74. https://doi.org/10.1016/j.ejmg.2016.01.008
[14] Lupo PJ, Schraw JM, Desrosiers TA, Nembhard WN, Langlois PH, Canfield MA, et al. Association between birth defects and cancer risk among children and adolescents in a population-based assessment of 10 million live births. JAMA Oncol. 2019;5(8):1150–8. https://doi.org/10.1002/gepi.1370120504
[15] Brockschmidt A, Chung B, Weber S, Fischer DC, Kolatsi-Joannou M, Christ L, et al. CHD1L: a new candidate gene for congenital anomalies of the kidneys and urinary tract (CAKUT). Nephrol Dialysis Transplant. 2012;27(6):2355–64. https://doi.org/10.1001/jamaoncol.2019.1215
[16] Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85(6):1429–33. https://doi.org/10.1093/ndt/gfr649
[17] Venizelos A, Elvebakken H, Perren A, Nikolaienko O, Deng W, Lothe IMB, et al. The molecular characteristics of high-grade gastroenteropancreatic neuroendocrine neoplasms. Endocrine-related Cancer. 2022;29(1):1–14. https://doi.org/10.1038/ki.2013.508
[18] Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–9. https://doi.org/10.1038/nm.3886
[19] Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30
[20] Nakken S, Saveliev V, Hofmann O, Moller P, Myklebost O, Hovig E. Cancer Predisposition Sequencing Reporter (CPSR): a flexible variant report engine for high-throughput germline screening in cancer. Int J Cancer. 2021;149(11):1955–60. https://doi.org/10.1038/gim.2015.30
[21] Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protocols. 2015;10(10):1556–66. https://doi.org/10.1038/nprot.2015.105
[22] Jones D, Raine KM, Davies H, Tarpey PS, Butler AP, Teague JW, et al. cgpCaVEManWrapper: simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS sata. Curr Protoc Bioinformatics. 2016;56:15.10.1–18. https://doi.org/10.1002/cpbi.20
[23] Raine KM, Hinton J, Butler AP, Teague JW, Davies H, Tarpey P, et al. cgpPindel: identifying somatically acquired insertion and deletion events from paired end sequencing. Curr Protoc Bioinformatics. 2015;52:15.7.1–2. https://doi.org/10.1002/0471250953.bi1507s52
[24] Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44(16):e131. https://doi.org/10.1093/nar/gkw520
[25] Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4(2):216–31. https://doi.org/10.1158/2159-8290.CD-13-0639
[26] Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, et al. From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci U S A. 2002;99(11):7518–23. https://doi.org/10.1073/pnas.102091699
[27] Muller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz-Erian P, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40(10):1163–5. https://doi.org/10.1038/ng.225
[28] Capasso M, Montella A, Tirelli M, Maiorino T, Cantalupo S, Iolascon A. Genetic predisposition to solid pediatric cancers. Front Oncol. 2020;10:590033. https://doi.org/10.1038/ng.225
[29] Gillani R, Camp SY, Han S, Jones JK, Chu H, O’Brien S, et al. Germline predisposition to pediatric Ewing sarcoma is characterized by inherited pathogenic variants in DNA damage repair genes. Am J Hum Genet. 2022;109(6):1026–37. https://doi.org/10.3389/fonc.2020.590033
[30] Roland JT, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR. Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci U S A. 2011;108(7):2789–94. https://doi.org/10.1016/j.ajhg.2022.04.007
[31] Akhavanfard S, Padmanabhan R, Yehia L, Cheng FX, Eng C. Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat Commun. 2020;11(1):2206. https://doi.org/10.1038/s41467-020-16067-1
[32] Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–46. https://doi.org/10.1056/NEJMoa1508054