ORIGINAL ARTICLE
Exploring the somatic mutational landscape of ovarian cancer in Estonia
Mikk Toominga,b  , Kadri Rekkerb , Kadri Toomeb , Laura Rohtc , Piret Laidrec, Olga Fjodorovab , Ustina Šamarinab , Sander Pajusalua,b , Mihkel Ilissona,b , Pilleriin Peetsd , Tõnu Jõgie, Karin Klaupe, Kristiina Ojamaaf , Eva-Maria Niine-Roolahtf, Piret Kaardef, Elen Vettusg , Jüri Piirsooh, Helen Vaharh, Katrin Õunapa,b and Tiina Kahrea,b
, Kadri Rekkerb , Kadri Toomeb , Laura Rohtc , Piret Laidrec, Olga Fjodorovab , Ustina Šamarinab , Sander Pajusalua,b , Mihkel Ilissona,b , Pilleriin Peetsd , Tõnu Jõgie, Karin Klaupe, Kristiina Ojamaaf , Eva-Maria Niine-Roolahtf, Piret Kaardef, Elen Vettusg , Jüri Piirsooh, Helen Vaharh, Katrin Õunapa,b and Tiina Kahrea,b
aDepartment of Genetics and Personalized Medicine, Institute of Clinical Medicine, University of Tartu, Tartu, Estonia; bDepartment of Laboratory Genetics, Genetics and Personalized Medicine Clinic, Tartu University Hospital, Tartu, Estonia; cDepartment of Clinical Genetics, Genetics and Personalized Medicine Clinic, Tartu University Hospital, Tartu, Estonia; dChair of Analytical Chemistry, Institute of Chemistry, University of Tartu, Tartu, Estonia; eHaematology and Oncology Clinic, Tartu University Hospital, Tartu, Estonia; fOncology and Haematology Clinic, North Estonian Medical Center, Tallinn, Estonia; gCentre of Oncology, East Tallinn Central Hospital, Tallinn, Estonia; hGynaecology Department, West Tallinn Central Hospital, Tallinn, Estonia
ABSTRACT
Background and purpose: This retrospective study provides Estonia’s first nationwide overview of pathogenic and likely pathogenic variants in ovarian cancer (OC) using routine tumor molecular profiling. The aim was to characterize the mutational landscape, identify clinically actionable alterations, and evaluate the integration of germline testing.
Patient/material and methods: A total of 339 OC tumor samples underwent next‑generation sequencing-based profiling. Clinical characteristics, histological subtype, and prior germline testing were recorded. Variants were classified according to clinical‑actionability levels, and homologous recombination deficiency (HRD) was assessed in high‑grade serous OC cases.
Results: The mean age at diagnosis was 62.4 ± 12.5 years, and high‑grade serous OC accounted for 82.0% of cases. Clinically actionable alterations (OncoKB Level 1&2) were detected in 37.5% of patients, most frequently involving BRCA1, BRCA2, KRAS, and BRAF. Additional therapeutic targets were identified across diverse biomarkers. HRD was present in 53.2% of tested high‑grade serous tumors. Germline testing had been performed in 41.9% of patients, revealing pathogenic or likely pathogenic variants in 22.5%, predominantly in BRCA1 and BRCA2.
Interpretation: This study outlines the mutational spectrum of OC in Estonia and demonstrates a substantial prevalence of actionable alterations and HRD. The findings highlight the value of comprehensive tumor profiling to support precision‑medicine approaches and improve individualized patient management in Estonia.
KEYWORDS: ovarian neoplasms; high-throughput nucleotide sequencing; genetic testing
Citation: ACTA ONCOLOGICA 2026, VOL. 65, 422–429. https://doi.org/10.2340/1651-226X.2026.45612.
Copyright: © 2026 The Author(s). Published by MJS Publishing on behalf of Acta Oncologica. This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Received: 24 February 2026; Accepted: 24 April 2026; Published: 18 May 2026
CONTACT: Mikk Tooming mikk.tooming@kliinikum.ee L. Puusepa 2, Tartu 50406, Estonia
Supplemental data for this article can be accessed online at https://doi.org/10.2340/1651-226X.2026.45612
Competing interests and funding: The authors report there are no competing interests to declare.
Introduction
According to the World Health Organization (WHO), ovarian cancer (OC) is the seventh most common cancer in women [1]. In Estonia, on average, ~140 people develop OC per year, according to the Estonian National Institute of Health Development, Health Statistics and Health Research Database [2]. The majority of newly diagnosed OC patients present with advanced-stage disease, which can contribute to poor survival outcomes despite advances in surgical and systemic treatment [3]. Cancer development is a complex, multistep process driven by pathogenic/likely pathogenic (PV) in genes that regulate cell growth and/or DNA repair mechanisms [4, 5]. These PV can be of germline or somatic origin, and understanding their contribution is essential for elucidating the mechanisms of inherited and acquired diseases [5]. Current clinical guidelines recommend offering genetic testing (GT) for risk evaluation to all women diagnosed with OC [5, 6]. Identifying germline PV can also prompt cascade screening for at-risk relatives. Germline variants, inherited in an autosomal dominant pattern [7], markedly increase the lifetime risk of developing specific cancers, frequently manifesting at an early age. For example, carriers of germline BRCA1 PV have an estimated 58% lifetime risk of OC, while BRCA2 carriers have a cumulative risk of approximately 29% [5]. Generally, germline GT has revealed PV in the BRCA1 and BRCA2 genes in up to 15% of women diagnosed with OC, highlighting a significant hereditary component [8–13]. Additionally, somatic PV in these same genes are identified in approximately 7% of cases, further emphasizing the importance of comprehensive molecular profiling in guiding personalized treatment strategies [6, 8]. Somatic mutation testing has emerged as a cornerstone of precision oncology in managing OC, offering critical insights into tumor biology and guiding personalized therapeutic strategies [6]. Somatic testing can also detect PV that can be of germline origin. Routine practice now favors somatic tumor testing in all advanced OC [6, 8, 14]. Historically, the widespread adoption of comprehensive tumor profiling was limited by therapeutic strategies, high sequencing costs and technological constraints [10, 13, 15]. Formalin-fixed paraffin-embedded (FFPE) tissue remains the most widely available and routinely archived material for molecular testing in OC [16]. At the same time, FFPE-derived DNA is often fragmented and chemically modified, which compromises the accuracy of downstream applications. However, the development of targeted next-generation sequencing (NGS) using comprehensive gene panels has provided a cost-effective and scalable solution [6, 17]. These advances have enabled FFPE-based NGS integration into routine clinical practice, supporting precision oncology in OC.
High-grade serous ovarian cancer (HGSOC) represents the most prevalent subtype of epithelial OC, accounting for the majority of cases and associated with poor prognosis due to its aggressive nature and frequent late-stage diagnosis [4, 18, 19]. Given the heterogeneous mutation spectrum in OC, broad tumor profiling panels can identify established driver genes and emerging tumor-specific biomarkers. These include homologous recombination deficiency (HRD) [6], microsatellite instability (MSI) [8], and tumor mutation burden (TMB) [20, 21], all of which can inform therapeutic decisions. Approximately 50% of HGSOC cases exhibit HRD [3, 6, 22]. HRD manifests through characteristic genomic alterations, including large-scale transitions, loss of heterozygosity, and telomeric allelic imbalance that reflect impaired homologous recombination repair beyond detectable PV in canonical HR genes. As such, HRD serves as a surrogate biomarker for predicting poly-ADP ribose polymerase (PARP) inhibitor sensitivity [6, 22, 23]. The OC treatment landscape has evolved with PARP inhibitors showing established clinical benefit, and emerging evidence suggests potential benefit from immunotherapies [6, 22, 24, 25]. Integration of tumor sequencing results with clinical parameters, such as chemotherapy response, progression-free survival, and tumor characteristics, can refine prognostic accuracy and support more optimized treatment selection [11]. It has been demonstrated that tumor DNA testing for BRCA1, BRCA2, and other relevant molecular markers provides additional value even when germline GT has already been performed, as tumor GT can uncover somatic alterations that are missed by germline analysis [8, 12]. This work provides the most comprehensive overview of actionable PV and biomarker profiles in Estonia, supporting the integration of precision medicine into routine oncology across the national healthcare system, and offering additional value through its combined somatic and germline testing perspective.
Patients/material and methods
Participants
The OC tumor samples underwent comprehensive genomic profiling at the Genetics and Personalized Medicine Clinic of Tartu University Hospital (TUH) between 2020 and 2024. As TUH is the only accredited center in Estonia performing OC molecular profiling, this represents a nationwide cohort, and the reported numbers reflect OC samples analyzed in the country during the study period. In total, 339 tumor samples were profiled, of which three were excluded due to insufficient DNA quality. Clinical data, including age at diagnosis, tumor cell fraction, histological subtype, and results of germline testing, were retrospectively obtained from test requisition forms completed by the ordering clinicians. Patients diagnosed with OC but not referred for molecular testing were not included, as molecular data were unavailable for these cases.
OC GT approaches during the study period
Until 2020, OC GT was primarily employed through germline-targeted testing approaches [30]. Somatic tumor testing was introduced in 2020 using TruSight Oncology 500 (523 genes; Illumina Inc.). Since 2022, OC testing has primarily followed a tumor-first GT strategy, with possible germline GT performed via Sanger seuqencing or targeted germline NGS panels. At the beginning of 2023, TSO500 RNA oligos were replaced with TruSight RNA Pan-Cancer oligos, and the add-on oligos of TruSight Oncology 500 HRD were introduced in July 2023 for HRD testing (Illumina, Inc.). Library preparation and sequencing were performed on the Illumina NextSeq 500 platform using 2×101bp paired-end reads, following the manufacturer’s protocols (Figure 1).
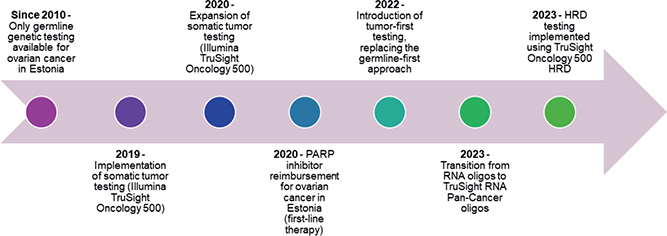
Figure 1. Timeline of ovarian cancer genomic testing and related clinical implementations in Estonia.
Tumor next-generation sequencing data analysis and data interpretation
Data were analyzed using the Illumina TSO500 Local App software versions 2.0–2.2 and from 2023, Illumina Dragen V4 server with DRAGEN TruSight Oncology 500 Analysis Software v.2.1.1 to v.2.5.3 (Illumina, Inc.). The filtering and annotation of identified genetic variants were performed using bcftools and variant effect predictor (VEP) software. For variant annotation, databases such as OncoKB, cBioPortal, VarSome, COSMIC, Franklin, MyCancerGenome, dbSNP, gnomAd, and ClinVar were utilized, along with predictive tools for protein function assessment, including Sorting Intolerant From Tolerant (SIFT), PolyPhen2, Combined Annotation-Dependent Depletion (CADD), and PhyloP. Variant classification followed American College of Medical Genetics (ACMG) guidelines [26], and variants were described according to the HGVS Nomenclature [27]. Only class 4 and 5 variants, as classified according to ACMG/Association of Molecular Pathology (AMP) guidelines, were reported in the clinical workflow [26]. Cutoff values for genomic biomarkers were defined as follows: single-nucleotide variants (SNV) were reported at a variant allele frequency (VAF) ≥ 5%; gene amplifications were considered significant at ≥ 5 copies, the applied method does not differentiate between focal amplifications and larger-scale copy-number increases across the locus. TMB-HIGH was classified as high at ≥ 10 mutations per megabase (mut/Mb); MSI-HIGH was defined as ≥ 20% unstable MSI sites; and HRD was considered positive in HGSOC when % VAF was ≥ 30 and the HRD score was ≥ 42, based on the TruSight Oncology 500 HRD assay aligned with the Myriad MyChoice methodology.
Results
Cohort characterization
This study employed comprehensive molecular profiling in routine clinical practice to analyze the mutational landscape and actionable oncogenic variants in OC tumor samples in Estonia. A total of 339 tumor samples from distinct individuals were included. The number of additional material requests was not recorded. The mean age at diagnosis for OC patients was 62.4 ± 12.5 years, while the mean age at comprehensive tumor profiling was 63.5 ± 12.4 years. The number of tumor samples tested increased threefold over the years, from 38 samples in 2020 to 114 samples in 2024.
Tumor characteristics
Most OC tumors were of primary origin, comprising 221 cases (65.2%), metastatic tumors accounted for 104 (30.7%) and unknown origin was reported in 14 samples (4.1%). HGSOC was the predominant histological subtype evaluated by pathologists, observed in 278 cases (82.0%), while low-grade serous ovarian cancer (LGSOC) represented 20 cases (5.9%). Other OC combined histologies accounted for 33 cases (9.7%), and an unknown histological type was reported in eight cases (2.4%). The mean pathologist‑estimated tumor fraction for OC samples was ~57.0% ± 24.0.
Sequencing metrics
The average sequencing coverage across all samples was 1171.6x(± 362.0), reflecting a high sequencing depth. Furthermore, the proportion of bases covered at 100x averaged 97.9% (± 1.0%).
Molecular findings in OC tumor samples
Among the 336 samples, we identified 529 single nucleotide PV. However, 14 samples harbored no PV. Among the detected PV, TP53 was the most frequently altered gene, present in 79.5% of cases. BRCA1 PV were the second most common (14.9%), followed by KRAS (6.0%), BRCA2 (5.7%), and PIK3CA (5.1%). In the HGSOC subtype, the most prevalent PV were TP53 87.4% of cases, BRCA1 16.2% of cases and BRCA2 (6.1%). In the other OC subtypes, the most prevalent PV were TP53 (41.4%), followed by a markedly higher occurrence of KRAS (20.7%), ARID1A (17.2%), PIK3CA (13.8%), and CTNNB1 (12.1%). The complete list of genes where PV was detected is shown Table 1. The most frequent BRCA1 PV were c.5266dup, detected in 12 of 336 individuals (3.6%), and c.4035del, detected in 10 of 336 individuals (3.0%). All PV by HGVS are listed in the additional information file ‘allPVs’.
Regarding the detected gene amplification events, PIK3CA was the most frequently amplified gene, observed in 34.2% of cases (Supplement table S1). Among the whole gene deletions identified, STK11 was the most frequent, accounting for 5.4% of cases (Supplement table S2). In the studied cohort, we identified two AR-V7 splicing alterations. Among the identified gene fusions, the most prevalent involved the ESR1 gene, accounting for eight events (2.4%) (Supplement table S3).
Tumor biomarker results
Among the 336 samples evaluated for TMB, only 13 (3.9%) reported as TMB-HIGH. MSI analysis revealed a similarly low instability frequency, with MSI-HIGH status detected in nine (2.7%) cases. HRD analysis was implemented in 2023 as part of our comprehensive tumor testing regimen in clinical practice. HRD analysis was performed in 94 of HGSOC, and HRD+ was reported in 50 cases (53.2%) of HGSOC.
Actionable variants were evaluated using the OncoKB database, focusing on Level 1 and 2 evidence for OC. Among OC‑specific actionable alterations, 126 individuals (37.5%) carried at least one actionable variant. The most frequently altered actionable gene was BRCA1 (14.6%), followed by KRAS (6.0%). Additional actionable findings are detailed in Table 2. When considering actionable biomarkers relevant to solid tumors more broadly, 56.3% of patients in our cohort had at least one targetable alteration (Supplement Table S4).
Germline diagnostic strategies employed on OC individuals
Among the individuals whose tumors were included in the somatic testing cohort, 142 (41.9%) had also undergone germline GT. From the overall germline GT results, PV was identified in 32 out of 142 (22.5%) individuals. The most frequently affected gene was BRCA1, with 16 carriers (11.3%), followed by BRCA2 in seven individuals (4.9%). The main BRCA1 PVs were c.4035del and c.5266dup, accounting for 68.8% of all germline BRCA1 PVs and 34.4% of all identified PVs. The primary BRCA2 PV identified was c.8572C>T, accounting for 28.6% of all BRCA2 germline PV. Additional germline PV were detected in CHEK2 in three patients (2.1%) and in BRIP1 in two patients (1.4%). Among the reported germline PV, one (3.2%) was not detected in the corresponding tumor tissue, a duplication in the RAD51C gene, encompassing exons 6 and 7. No germline PV were identified in 110 individuals (77.5%). All detected germline PV are shown in Supplement information ‘AllPVs’.
Discussion and conclusion
This nationwide retrospective analysis of 339 OC tumor samples demonstrates the feasibility and clinical value of implementing comprehensive molecular profiling within routine diagnostics in Estonia. The number of tested tumors increased steadily from 2020 to 2024, representing a threefold increase and illustrating the adoption of precision oncology practices across Estonian oncology centers. The mean age at OC diagnosis in our cohort was 62.4 years comparable to or slightly higher than onset ages reported in the USA (63.0), Croatia (59.0), and Tunisia (52.4) [18, 28, 29]. In Estonia prior to 2020, clinical practice relied primarily on germline GT. Somatic tumor profiling was introduced later and was ordered selectively for previously germline-tested patients when additional tumor-level information was needed for therapeutic decision-making. Following the reimbursement of PARP inhibitors such as olaparib in 2020 in first-line maintenance treatment, Estonia introduced a tumor‑first GT approach in 2022, with optional germline testing, alongside a marked increase in testing volumes (Figure 1). However, in our cohort, the median time from diagnosis to somatic testing was 1.1 years, whereas the median time to germline testing was substantially longer, 3.3 years [30]. This indicates that the implementation of somatic testing has, in fact, improved the timeliness of molecular evaluation in Estonia, as patients are now receiving at least somatic testing earlier than they historically received germline testing.
Recent Nordic and European initiatives further highlight the growing momentum toward precision cancer medicine. Large-scale collaborative programs such as PCM4EU and PRIME-ROSE have demonstrated the importance of coordinated precision medicine implementation across Europe to ensure equitable access to molecularly guided therapies, a development in which Estonia is also actively participating. Similar national initiatives in Norway and Finland show how structured precision medicine programs can rapidly increase patient inclusion, improve diagnostic workflows, and generate real-world evidence to support clinical decision-making. Nordic experts also emphasize that while precision medicine holds great promise, its successful integration into routine oncology requires sustained investment, interdisciplinary collaboration, and continuous evaluation of clinical impact [31].
Only three tumor samples (0.9%) failed quality control due to insufficient genomic material. Information regarding additional requests for FFPE material for nucleic acid extraction was not available. This failure rate is notably lower than those reported in previous studies, including 2.9% in a Japanese cohort and 6.1% in a multicenter study by Jantus-Lewintre et al., conducted across Austria, the Netherlands, Italy, France, and Spain [32, 33]. Centralized molecular profiling at TUH ensured high technical reliability, achieving a mean sequencing depth of 1171× and 97.9% of bases covered at ≥ 100× across all samples. These parameters enabled robust detection of somatic variants in FFPE-derived tumors and surpassed sequencing coverage reported in studies from Greece (500×), Croatia (500×; ≥ 100× > 99%), and a multicenter study by Jantus-Lewintre et al. (500×) [11, 28, 33]. This high level of sequencing coverage is critical for ensuring accurate variant calling in clinical cancer genomic profiling workflows.
TP53 PV are the hallmark alteration of HGSOC, present in over half of cases and including rare in-frame insertions, deletions, and frameshift mutations [28, 29, 34, 35]. Their prognostic significance remains unclear, though in vitro data suggest potential effects on PARP-inhibitor response [36]. In mucinous OC and LGSOC, PV in KRAS, BRAF, and NRAS are key drivers that converge on aberrant MAPK-pathway activation. Because KRAS-driven MAPK signaling underlies the pathogenesis of these subtypes, MEK-targeted therapy, as well as emerging EGFR-directed and combined MAPK/PI3K-pathway strategies, represent the most rational therapeutic approaches [34, 35, 37–39].
BRCA1 and BRCA2 PV occurred in 14.9% and 5.7% of tumors, which is well within the expected range for combined somatic and germline BRCA-associated OC (~15–20%) [12, 29]. Both previously known BRCA1 main PV in Estonia (c.5266dup and c.4035del) and the BRCA2 c.8572C>T PV were the most common alterations, mirroring the germline BRCA1/2 spectrum previously identified in the Estonian Biobank, regional cohorts [28, 40–43], and published data from the Estonian OC cohort by Tooming et al., where c.5266dup and c.4035del germline PV were found in 11.7% of 759 OC patients [30]. Importantly, in one patient, germline PV was not detected in the corresponding tumor, demonstrating that tumor-only analysis may fail to identify a subset of heritable variants. Guidelines from the European Society for Medical Oncology and the American Society of Clinical Oncology indicate that tumor-only sequencing may miss approximately 5% of germline BRCA1/2 PV [8, 14], emphasizing the clinical importance of paired tumor-germline testing for accurate risk assessment and cascade testing.
Biomarker analysis revealed low frequencies of TMB-HIGH (3.9%) and MSI-HIGH tumors (2.7%), consistent with reported rates in OC by Morand et al. (TMB-HIGH 3.6%) and Landen et al. (TMB-HIGH 3%; MSI-HIGH 0.3%) [24, 44]. As expected, HRD+ was detected in 53.2% of HGSOC, aligning with the ~50% prevalence reported in the literature [3, 6, 22]. However, HRD evaluation is technically complex, requiring a sufficiently high pathologist-assessed tumor cell fraction (30%) and reliable estimation for genomic instability metrics. As a result, only samples that fulfilled these criteria were included in the HRD analysis. Conversely, since HRD testing became part of routine tumor profiling in 2023, it is expected to play an increasingly important role in guiding treatment decisions for OC patients in Estonia.
Actionable variants defined by OncoKB Level 1& 2 in OC evidence were identified in 37.5% of patients, and this proportion is consistent with other national OC genomic profiling studies (~> 20%) [11, 13, 28, 29, 34], underscoring the clinical value of comprehensive tumor sequencing. The strongest evidence exists for BRCA1/2 and HRD‑associated alterations, which underpin the approved use of PARP inhibitors in OC. U.S. Food and Drug Administration (FDA)‑approved PARP inhibitor options are now largely restricted to the maintenance setting [9, 13, 22]. These indications closely align with European Medicines Agency (EMA) practice and remain most applicable to the HGSOC subgroup, where HRD and BRCA PV are concentrated [45]. Beyond HRD, our results identified additional, potentially druggable alterations. KRAS mutations, which are particularly enriched in non-HGSOC tumors such as LGSOC [46], have recently gained direct therapeutic relevance following the accelerated FDA approval (May 2025) of the avutometinib + defactinib combination for KRAS-mutated recurrent LGSOC. As these agents have not yet received EMA approval, patients in Europe currently require access through clinical trial participation or structured drug-repurposing pathways such as DRUP-type programs [15, 47]. Conversely, NTRK3 gene fusions are directly actionable through the tumor-agnostic FDA and EMA approvals of larotrectinib and entrectinib, making these alterations immediately clinically relevant for patients identified with such fusions. In addition, several variants identified in ERBB2, MET, FGFR2, and components of the PI3K/mTOR pathway represent additional potentially targetable alterations. Although these biomarkers have received FDA and EMA approvals for targeted therapies in other solid tumors, they are not yet specifically approved for OC. Nevertheless, clinical trials, such as the DESTINY-PanTumor02 study, are actively evaluating their therapeutic relevance in this setting [48]. These alterations may provide eligibility for tumor‑agnostic basket trials evaluating targeted strategies across cancer types. In such cases, patients may also qualify for precision‑oncology trials or compassionate‑use and/or drug‑repurposing programs, including DRUP‑type frameworks [15, 47].
The absence of longitudinal clinical outcome data limited our ability to assess the clinical relevance of the detected alterations. Additionally, germline testing was inconsistently performed, some patients had undergone germline analysis prior to somatic testing, whereas others had not undergone germline testing at all constraining our capacity to systematically evaluate germline somatic relationships. Future studies should integrate comprehensive genomic profiling with complete clinical datasets, including standardized germline testing, to enable robust outcome-based analyses and further advance precision oncology.
Collectively, these findings demonstrate that integrating comprehensive tumor genomic profiling with germline GT substantially enhances diagnostic yield and therapeutic precision for OC patients in Estonia. The high prevalence of actionable biomarkers, particularly PV BRCA1/2 and HRD, supports the continued expansion of molecular diagnostics and integrative precision oncology frameworks. Although paired tumor-germline testing represents the optimal GT strategy, its routine adoption may be challenging due to cost or logistical constraints [49]. Expanding access where feasible, integrating HRD assessment, and improving clinical data linkage remain key to advancing OC precision care in Estonia.
Acknowledgements
We thank all the patients, and all the people involved in this work. This work could not have been accomplished without the daily efforts of colleagues in the Genetics and Personalized Medicine Clinic at TUH, as well as oncologists and pathologists from TUH, North Estonian Medical Center, East Tallinn Central Hospital, and West Tallinn Central Hospital. The Estonian Research Council supported this work through grants PSG774, PSG1027 and PRG2040.
Data availability statement
The genomic data generated in this study originate from accredited clinical diagnostic testing at TUH. Due to institutional policy, patient consent restrictions, and national data protection regulations, raw sequencing files and individual level genetic data cannot be deposited in public repositories. De-identified aggregate data supporting the findings of this study are included within the article and its supplementary materials. Access to controlled level clinical genomic data may be granted on reasonable request for research purposes that comply with applicable regulations. Requests should be directed to Mikk Tooming (mikk.tooming@kliinikum.ee).
Ethics declarations & trial registry information
This study is a hospital quality research project, authorized by TUH and the TUH Clinical Research Centre and has valid approval IDs by Research Ethics Committee of the University of Tartu (332/T-2, 21.12.2020; 341/M-2, 17.02.2021; 359/M-9, 21.02.2022; 384/M-14, 20.11.2023; 389/M-1, 15.04.2024; 406/M-6, 25.11.2025). This was a non-experimental study, and no new biological samples were collected for its purposes. The study analyzed data from patients tested at the TUH, Genetics and Personalized Medicine Clinic, Molecular Diagnostic Laboratory, as part of the routine clinical workflow in compliance with ISO 15189:2022 standards and under the Estonian Accreditation Centre license (M005). All methods were carried out in accordance with relevant guidelines and regulations.
Author contributions
M.T. served as the primary author of the manuscript, collected the data, and contributed to the interpretation of the results. M.T., together with T.K. and K.Õ., analyzed the results in collaboration with the other authors. T.K. and K.Õ., as senior advisors, oversaw the analysis of results, tables, and figures, and contributed to manuscript writing. M.T., K.R., K.T., O.F., and T.K. contributed to the interpretation of genetic results. U.Š. performed the bioinformatics analyses. L.R., P.L., T.J., K.K., K.O., E.N., P.K., E.V., J.P., and H.V. were involved in patient data collection. M.I. and P.P. performed the statistical analyses in collaboration with M.T. All authors reviewed and edited the manuscript and approved the final version.
References
[1] WHO. WHO, cancer today [Internet]. 2026 [cited 2026 Feb 21]. Available from: https://gco.iarc.who.int/today/
[2] Tervisestatistika ja terviseuuringute andmebaas. PK10: Pahaloomuliste kasvajate esmasjuhud paikme, soo ja vanuserühma järgi [Internet]. [cited 2025 Apr 14]. Available from: http://statistika.tai.ee/pxweb/et/Andmebaas/Andmebaas__02Haigestumus__04PahaloomulisedKasvajad/PK10.px/
[3] Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. https://doi.org/10.1038/nature10166
[4] Shih IM, Wang Y, Wang TL. The origin of ovarian cancer species and precancerous landscape. Am J Pathol. 2021;191(1):26–39. https://doi.org/10.1016/j.ajpath.2020.09.006
[5] Daly MB, Pal T, Maxwell KN, Churpek J, Kohlmann W, AlHilli Z, et al. NCCN guidelines® insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 3.2026. J Natl Compr Canc Netw [Internet]. 2026;(v3.2026). [cited 2026 Feb 2]. Available from: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bopp.pdf
[6] Gressel GM, Frey MK, Norquist B, Senter L, Blank SV, Urban RR. Germline and somatic testing for ovarian cancer: an SGO clinical practice statement. Gynecol Oncol. 2024;181:170–8. https://doi.org/10.1016/j.ygyno.2023.12.010
[7] Casaubon JT, Kashyap S, Regan JP. BRCA1 and BRCA2 mutations [Internet]. Treasure Island, FL: StatPearls Publishing; 2025 [cited 2026 Feb 21]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK470239/
[8] Konstantinopoulos PA, Norquist B, Lacchetti C, Armstrong D, Grisham RN, Goodfellow PJ, et al. Germline and somatic tumor testing in epithelial ovarian cancer: ASCO guideline. J Clin Oncol. 2020;38(11):1222–45. https://doi.org/10.1200/JCO.19.02960
[9] Barbosa A, Pinto P, Peixoto A, Guerra J, Pinto C, Santos C, et al. Gene panel tumor testing in ovarian cancer patients significantly increases the yield of clinically actionable germline variants beyond BRCA1/BRCA2. Cancers. 2020;12(10):2834. https://doi.org/10.3390/cancers12102834
[10] Jang J, Kim Y, Kim JH, Cho SM, Lee KA. Cost-effectiveness analysis of germline and somatic BRCA testing in patients with advanced ovarian cancer. Ann Lab Med. 2023;43(1):73–81. https://doi.org/10.3343/alm.2023.43.1.73
[11] Andrikopoulou A, Zografos E, Apostolidou K, Kyriazoglou A, Papatheodoridi AM, Kaparelou M, et al. Germline and somatic variants in ovarian carcinoma: a next-generation sequencing (NGS) analysis. Front Oncol. 2022;12:1030786. https://doi.org/10.3389/fonc.2022.1030786
[12] Frugtniet B, Morgan S, Murray A, Palmer-Smith S, White R, Jones R, et al. The detection of germline and somatic BRCA1/2 genetic variants through parallel testing of patients with high-grade serous ovarian cancer: a national retrospective audit. BJOG. 2022;129(3):433–42. https://doi.org/10.1111/1471-0528.16975
[13] Vos JR, Fakkert IE, de Hullu JA, van Altena AM, Sie AS, Ouchene H, et al. Universal tumor DNA BRCA1/2 testing of ovarian cancer: prescreening PARPi treatment and genetic predisposition. J Natl Cancer Inst. 2020;112(2):161–9. https://doi.org/10.1093/jnci/djz080
[14] Mosele MF, Westphalen CB, Stenzinger A, Barlesi F, Bayle A, Bièche I, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with advanced cancer in 2024: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2024;35(7):588–606. https://doi.org/10.1016/j.annonc.2024.04.005
[15] Tavares V, Marques IS, de Melo IG, Assis J, Pereira D, Medeiros R. Paradigm shift: a comprehensive review of ovarian cancer management in an era of advancements. Int J Mol Sci. 2024;25(3):1845. https://doi.org/10.3390/ijms25031845
[16] Carrick DM, Mehaffey MG, Sachs MC, Altekruse S, Camalier C, Chuaqui R, et al. Robustness of next generation sequencing on older formalin-fixed paraffin-embedded tissue. PLoS One. 2015;10(7):e0127353. https://doi.org/10.1371/journal.pone.0127353
[17] Edsjö A, Russnes HG, Lehtiö J, Tamborero D, Hovig E, Stenzinger A, et al. High-throughput molecular assays for inclusion in personalised oncology trials – state-of-the-art and beyond. J Intern Med. 2024;295(6):785–803. https://doi.org/10.1111/joim.13785
[18] Caruso G, Weroha SJ, Cliby W. Ovarian cancer: a review. JAMA. 2025;334(14):1278–91. https://doi.org/10.1001/jama.2025.9495
[19] Giro A, Herrmann T, Bauer A, Pinard C, Godiveau M, Passildas J, et al. Predictive and prognostic factors in epithelial ovarian cancer: a review. Indian J Gynecol Oncolog. 2025;23(1):45. https://doi.org/10.1007/s40944-025-00964-8
[20] Nero C, Ciccarone F, Pietragalla A, Duranti S, Daniele G, Salutari V, et al. Ovarian cancer treatments strategy: focus on PARP inhibitors and immune check point inhibitors. Cancers. 2021;13(6):1298. https://doi.org/10.3390/cancers13061298
[21] Cui M, Xia Q, Zhang X, Yan W, Meng D, Xie S, et al. Development and validation of a tumor mutation burden-related immune prognostic signature for ovarian cancers. Front Genet. 2022;12:688207. https://doi.org/10.3389/fgene.2021.688207
[22] O’Malley DM, Krivak TC, Kabil N, Munley J, Moore KN. PARP inhibitors in ovarian cancer: a review. Target Oncol. 2023;18(4):471–503. https://doi.org/10.1007/s11523-023-00970-w
[23] Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Canc Res. 2014;16(3):211. https://doi.org/10.1186/bcr3670
[24] Morand S, Devanaboyina M, Staats H, Stanbery L, Nemunaitis J. Ovarian cancer immunotherapy and personalized medicine. Int J Mol Sci. 2021;22(12):6532. https://doi.org/10.3390/ijms22126532
[25] Colombo N, Coleman RL, Wu X, Köse F, Wenham RM, Sebastianelli A, et al. 2022-RA-657-ESGO ENGOT-ov65/KEYNOTE-B96: phase 3, randomized, double-blind study of pembrolizumab versus placebo plus paclitaxel with optional bevacizumab for platinum-resistant recurrent ovarian cancer. Int J Gynecol Cancer. 2022;32:A251. https://doi.org/10.1136/ijgc-2022-ESGO.538
[26] Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23. https://doi.org/10.1038/gim.2015.30
[27] den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37(6):564–9. https://doi.org/10.1002/humu.22981
[28] Čerina D, Matković V, Katić K, Belac Lovasić I, Šeparović R, Canjko I, et al. Comprehensive genomic profiling in the management of ovarian cancer – national results from croatia. J Pers Med. 2022;12(7):1176. https://doi.org/10.3390/jpm12071176
[29] Ammous-Boukhris N, Abdelmaksoud-Dammak R, Ben Kridis W, Ben-Ayed-Guerfali D, Shtaiwi Abed A, Guidara S, et al. Germline and somatic mutational variants of Tunisian high grade serous ovarian cancer identified by next-generation sequencing. BMC Cancer. 2025;25:1542. https://doi.org/10.1186/s12885-025-14989-x
[30] Tooming M, Toome K, Rekker K, Roht L, Laidre P, Fjodorova O, et al. Exploring the hereditary genetic mutational landscape of breast and ovarian cancer in Estonia. Sci Rep. 2026;16(1):13373. Available from: https://www.nature.com/articles/s41598-026-43459-y doi: https://doi.org/10.1038/s41598-026-43459-y
[31] Nilbert M. The promises of precision medicine – voices from the Nordics. Acta Oncol. 2025;64:775–7. https://doi.org/10.2340/1651-226X.2025.43987
[32] Imoto K, Yamamoto H, Ohkawa C, Shimada N, Ikuzawa R, Takeda H, et al. An approach for improvement of the accuracy of cancer gene panel testing. Int J Clin Oncol. 2024;29(5):571–81. https://doi.org/10.1007/s10147-024-02483-6
[33] Jantus-Lewintre E, Rappa A, Ruano D, van Egmond D, Gallach S, Gozuyasli D, et al. Multicenter in-house evaluation of an amplicon-based next−generation sequencing panel for comprehensive molecular profiling. Mol Diagn Ther. 2025;29(2):249–61. https://doi.org/10.1007/s40291-024-00766-2
[34] Fieuws C, Van der Meulen J, Proesmans K, De Jaeghere EA, Loontiens S, Van Dorpe J, et al. Identification of potentially actionable genetic variants in epithelial ovarian cancer: a retrospective cohort study. npj Precis Onc. 2024;8(1):1–9. https://doi.org/10.1038/s41698-024-00565-2
[35] Veneziani AC, Gonzalez-Ochoa E, Alqaisi H, Madariaga A, Bhat G, Rouzbahman M, et al. Heterogeneity and treatment landscape of ovarian carcinoma. Nat Rev Clin Oncol. 2023;20(12):820–42. https://doi.org/10.1038/s41571-023-00819-1
[36] Smith LE, Padilla JL, Licor A, Steinkamp MP, Lagutina IV, Guo Y, et al. Novel p53 reactivators that are synergistic with olaparib for the treatment of gynecologic cancers with mutant p53. Transl Oncol. 2025;61:102522. https://doi.org/10.1016/j.tranon.2025.102522
[37] El-Bahrawy M. Ovarian cancer pathology. In: Farghaly SA, editor. Advances in diagnosis and management of ovarian cancer. Cham: Springer International Publishing; 2022. p. 57–85. https://doi.org/10.1007/978-3-031-09169-8_5
[38] Kelliher L, Yoeli-Bik R, Schweizer L, Lengyel E. Molecular changes driving low-grade serous ovarian cancer and implications for treatment. Int J Gynecol Cancer. 2024;34(10):1630–8. https://doi.org/10.1136/ijgc-2024-005305
[39] Shim YS, Kim JH, Seo S-S, Kang S, Park SY, Lim MC. Landscape of genomic alterations and clinical outcomes in low-grade serous ovarian cancer in Korea. Gynecol Oncol Rep. 2025;61:101934. https://doi.org/10.1016/j.gore.2025.101934
[40] Koczkowska M, Zuk M, Gorczynski A, Ratajska M, Lewandowska M, Biernat W, et al. Detection of somatic BRCA1/2 mutations in ovarian cancer – next‐generation sequencing analysis of 100 cases. Cancer Med. 2016;5(7):1640–6. https://doi.org/10.1002/cam4.748
[41] Leitsalu L, Palover M, Sikka TT, Reigo A, Kals M, Pärn K, et al. Genotype-first approach to the detection of hereditary breast and ovarian cancer risk, and effects of risk disclosure to biobank participants. Eur J Hum Genet. 2021;29(3):471–81. https://doi.org/10.1038/s41431-020-00760-2
[42] Jürgens H, Roht L, Leitsalu L, Nõukas M, Palover M, Nikopensius T, et al. Precise, genotype-first breast cancer prevention: experience with transferring monogenic findings from a population biobank to the clinical setting. Front Genet. 2022;13:881100. https://doi.org/10.3389/fgene.2022.881100
[43] Tamboom K, Kaasik K, Aršavskaja J, Tekkel M, Lilleorg A, Padrik P, et al. BRCA1 mutations in women with familial or early-onset breast cancer and BRCA2 mutations in familial cancer in Estonia. Hered Cancer Clin Pract. 2010;8(1):4. https://doi.org/10.1186/1897-4287-8-4
[44] Landen CN, Molinero L, Hamidi H, Sehouli J, Miller A, Moore KN, et al. Influence of genomic landscape on cancer immunotherapy for newly diagnosed ovarian cancer: biomarker analyses from the IMagyn050 randomized clinical trial. Clin Cancer Res. 2023;29(9):1698–707. https://doi.org/10.1158/1078-0432.CCR-22-2032
[45] European Medicines Agency (EMA). European Medicines Agency (EMA) – olaparib [Internet]. 2026 [cited 2026 Mar 22]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/lynparza
[46] Peng Y, Yang Q. Targeting KRAS in gynecological malignancies. FASEB J. 2024;38(19):e70089. https://doi.org/10.1096/fj.202401734R
[47] Villegas-Vazquez EY, Marín-Carrasco FP, Reyes-Hernández OD, Báez-González AS, Bustamante-Montes LP, Padilla-Benavides T, et al. Revolutionizing ovarian cancer therapy by drug repositioning for accelerated and cost-effective treatments. Front Oncol. 2025;14:1514120. https://doi.org/10.3389/fonc.2024.1514120
[48] Meric-Bernstam F, Makker V, Oaknin A, Oh DY, Banerjee S, González-Martín A, et al. Efficacy and safety of trastuzumab deruxtecan in patients with HER2-expressing solid tumors: primary results from the DESTINY-PanTumor02 phase II trial. J Clin Oncol. 2024;42(1):47–58. https://doi.org/10.1200/JCO.23.02005
[49] Kuzbari Z, Bandlamudi C, Loveday C, Garrett A, Mehine M, George A, et al. Germline-focused analysis of tumour-detected variants in 49,264 cancer patients: ESMO Precision Medicine Working Group recommendations. Ann Oncol. 2023;34(3):215–27. https://doi.org/10.1016/j.annonc.2022.12.003